Practical: Introduction to quantum mechanics / molecular mechanics (QM/MM) simulations
Bert de Groot
This practical is a modification of a practical written by Gerrit Groenhof.
Contents
Introduction
In the previous lectures, you learned about quantum chemical methods
that allow one to compute the electronic structure of molecules. In
this practical, we will use these methods to study a chemical
reactions of a DNA molecule. To be more precise, we study the
dimerization of thymine within a DNA double strand.
Classical MD simulations, based on Newton's equations of motion and
classical force fields, are quite
efficient and allow simulations on
time scales up to microseconds or even longer.
A number of such simulations were conducted in the practicals of the
last semester. However, classical force field cannot describe the
formation or breaking of chemical bonds.
Thus, if we wish to study
chemical reactions, we must describe the molecular system
using quantum mechanical methods. Unfortunately, these are orders of
magnitude more expensive and scale unfavorably with the number of
atoms. As a consequence, we cannot describe a complete biomolecule
(protein, DNA, ...) quantum-mechanically.
Question:
Is the lack of bond-breaking a fundamental limitation of the use of classical force fields?
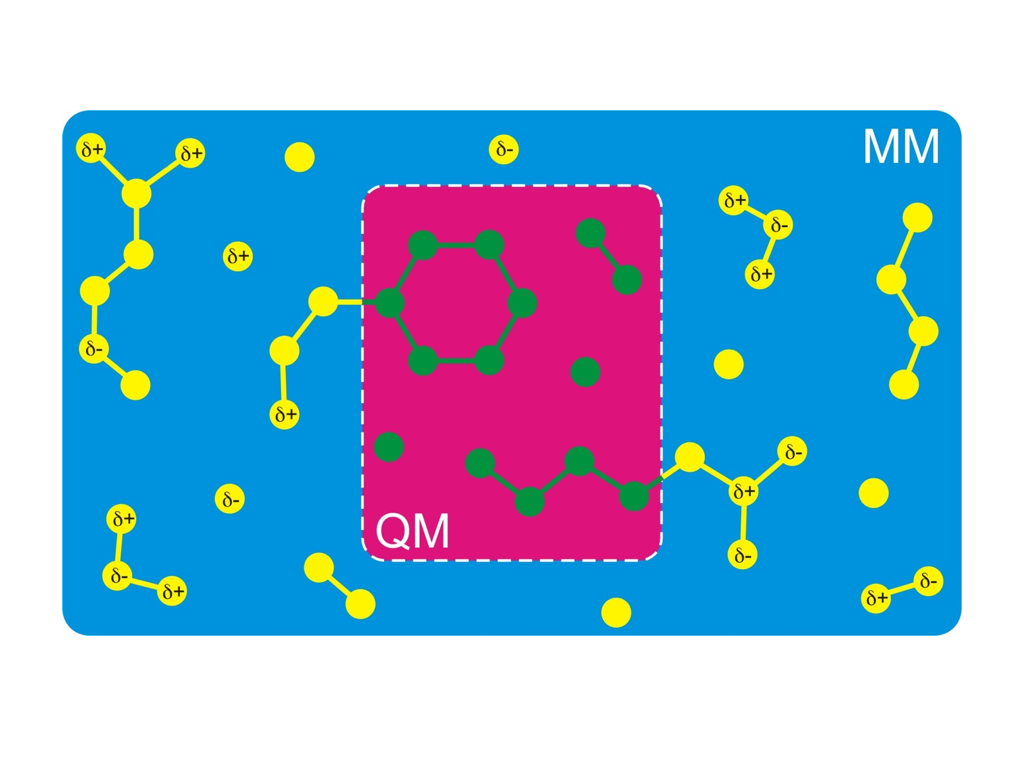
A simulation technique that nevertheless allows you to simulate the chemistry of
biomolecules is called hybrid QM/MM (quantum mechanics/molecular
mechanics). The idea is to treat only that part of the molecule
quantum mechanically, where the chemical reaction takes place (red
background in the figure on the right-hand side). The
rest is treated using a normal classical Newtonian dynamics and classical force field (blue background in
the figure). Notably, the Nobel Prize for Chemistry was awarded in 2013 to Martin Karplus, Michael Levitt,
and Arieh Warshel, in part for the development of the QM/MM method.
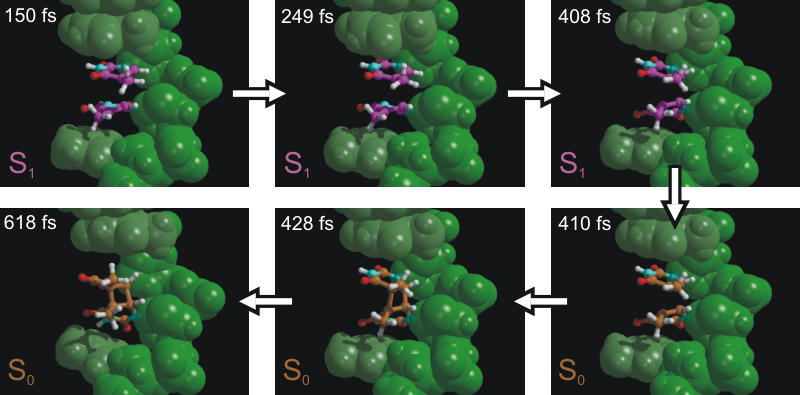
As mentioned above, we here study the dimerization of the nucleotide
thymine within one strand of a DNA double helix. Intra-strand thymine
dimerization (see figure above) is recognized as
the most common process leading to DNA damage under
ultraviolet (UV) irradiation. The formation of thymine dimers has
potentially important physiological consequences. This mutagenic
photoproduct can disrupt the function of DNA and thereby trigger
complex biological responses, including apoptosis (triggered cell death), immune
suppression, and carcinogenesis (development of cancer).
To survive the exposure to UV radiation, organisms have evolved complex
mechanisms to repair damaged DNA. The initial step is usually the
detection of a damage spot, a thymine dimer for
instance. Subsequently, the dimer is either repaired, or completely
removed.
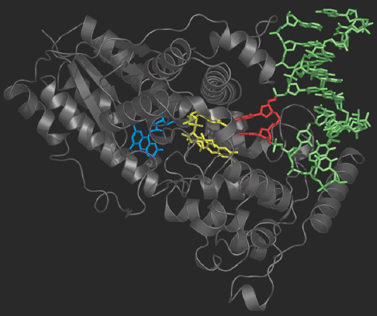
Photolyase is an enzyme that detects the dimer site by binding to it
and then catalyses the splitting of the dimer into the original
pyrimidine bases (figure on the right). Photolyase contains a reduced flavin
co-enzyme co-factor that upon absorption of UV light donates an
electron to the bound Thyime dimer. The excess electron destabilizes
the dimer, and facilitates the slitting of the cyclobutane ring. After
the original thymine bases are restored, the electron flows back onto
the flavin and Photolyase is ready to repair the next lesion.
Thymine dimers can also be restored without the help of Photolyase. In
this so-called self-repair process, the dimer splits upon spontaneous
uptake of an electron. Depending on the base sequence, such electrons
are readily available. In this tutorial we will examine the self
repair process by means of QM/MM simulations.
Go back to Contents
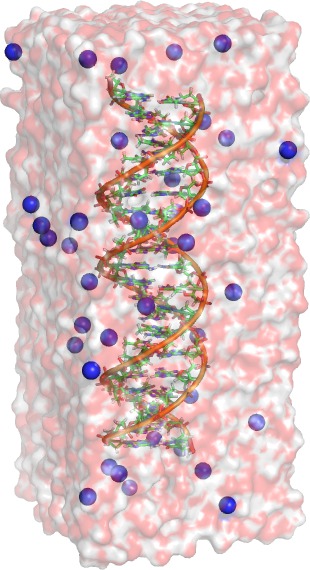
Setup of DNA simulation system
In the following, we set up a simulation system of a DNA double helix
solvated in water. A PDB structure of an helix and the required force
field files can be found here. Retrieve and unpack the gzipped tar ball
with
wget http://www3.mpibpc.mpg.de/groups/de_groot/compbio/p7/files/setup-helix.tar.gz
tar xvzf setup-helix.tar.gz
cd setup-helix
Take a look at the structure in your
favorite molecular viewer (PyMol, Rasmol, VMD). For VMD, run:
Zoom in and have a
closer look at the nucleotides. You will notice that one strand of the
helix contains only adenine and the other
only thymine.
The simulations will be carried out with the GROMACS simulation package. On the
GROMACS homepage you can find both
the software and documentation (online reference and paper manual).
To run a simulation, three input files are usually required:
- a structure file (.pdb), containing the atomic coordinates of the system
to be simulated
- a "molecular topology" (.top), containing the atomic simulation
parameters (force-field) and a description of the bonds, bond angles,
etc. of the simulation system
- the simulation parameters (.mdp): type of simulation, number of steps,
simulation temperature, etc.
Any GROMACS command is executed via a single Linux executable, named gmx. As shown below,
a specific GROMACS command gromacscommand is called via a call such as:
gmx gromacscommand options...
 First, we generate a topology with the Gromacs command
pdb2gmx. The file helix.pdb contains two special thymine
types with residue names DTa and DTb, respectively. pdb2gmx will read
the residue topology database amber99.ff/dna.rtp, in which two special
chemical bonds between DTa and DTb are defined. These two bonds make
the dimer. Run:
First, we generate a topology with the Gromacs command
pdb2gmx. The file helix.pdb contains two special thymine
types with residue names DTa and DTb, respectively. pdb2gmx will read
the residue topology database amber99.ff/dna.rtp, in which two special
chemical bonds between DTa and DTb are defined. These two bonds make
the dimer. Run:
gmx pdb2gmx -ignh -f helix.pdb -o conf.pdb
Choose the AMBER99 force field in the present working directory (option 1) and the TIP3P water model.
Please note that the commands in bold print can be easily transferred to the
command prompt with copy-and-paste (select text by dragging the mouse over it
with the left mouse button pressed, and paste by pressing the middle mouse
button).
Hint: ls -lrt prints the files in the current directory, with the most recent files at the bottom. So you can easily see which files were written by the last command.
Next, we place the structure written by pdb2gmx into a simulation box (using the editconf command), and we add water to the simulation box:
gmx editconf -c -d 1.0 -f conf.pdb -o boxed.pdb
and
gmx solvate -cs spc216.gro -cp boxed.pdb -p topol -o solved.pdb
Now, we neutralize the overall negative charge of -38 by adding 38
sodium ions. We first need to create a tpr file, for which we use neutralize.mdp. First, we need to
generate a tpr file using grompp,
gmx grompp -f neutralize.mdp -c solved.pdb -r solved.pdb -maxwarn 1
then we use the Gromacs command genion to add the ions:
gmx genion -np 38 -pname NA -o neutral.pdb -p
Choose 3 to tell genion that you want to replace water molecules by
sodium (Na) ions.
Take a look for instance in VMD (type vmd neutral.pdb) to have a look at the
system.
Now, the structure is in a state of high-energy because of two
reasons. (a) potential overlap between added water molecules and DNA;
(b) because the chemical bonds that connect the thymine monomers are
not relaxed. Therefore, we initiate our energy minimization using the
steepest descent algorithm.
gmx grompp -f steep.mdp -c neutral.pdb -r neutral.pdb
If Gromacs complains about a non-zero net charge, find the output line that
states the net charge. Might this just be a floating point truncation error?
If you are certain that this is the case, you can ignore the net-charge
warning by adding -maxwarn 1 to the command:
gmx grompp -f steep.mdp -c neutral.pdb -r neutral.pdb -maxwarn 1
Now carry out the steepest descent algorithm.
gmx mdrun -v -c neutral_mini.pdb
Have a look at the potential energy (Epot) in the mdrun output - does it decrease as expected?
Question:
What would happen in the simulation if we would not carry out an energy minimization beforehand? (Hint: have a look at the maximum force as well).
Finally, we equilibrate the compute simulation system for 10ps. Use
the MD parameter file equil.mdp
for that:
gmx grompp -f equil.mdp -c neutral_mini.pdb -r neutral_mini.pdb -maxwarn 1
Then carry out the equilibrium simulation:
gmx mdrun -v -c equil.pdb
This simulation may take a minute or two. You can also get the final
structure here. Take a look at the
equilibration of the DNA strand in VMD. Because we wrote only the DNA
atoms into the trajectory files traj.xtc, we first need a PDB
structure file with the DNA only (take out sodium NA and water SOL
from the PDB file). We use the egrep command for that purpose:
egrep -v ' NA | SOL' neutral_mini.pdb > neutral_mini_dna.pdb
Then use:
and observe the equilibration. Do the chemical bonds connecting the
two thymines look reasonable?
Hint: If something went wrong, you can also download the trajectory and the pdb files here: traj_comp.xtc
and neutral_mini_dna.pdb
Go back to Contents
First QM/MM simulation
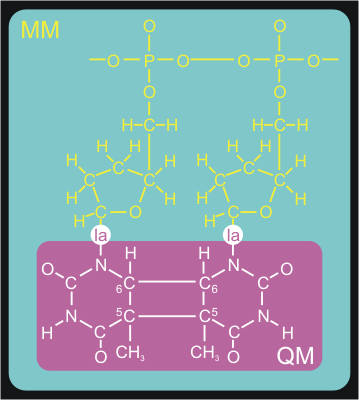
We want to set up the system for a QM/MM simulation with
Gromacs. The dimerized thymine bases will be described at the
semi-empirical AM1 level of theory, while the remainder of the system
is modeled with the Amber99 forcefield. Figure 3 shows how we split up
our system in a QM and MM part.
The QM/MM division splits the systems along a chemical
bond. Therefore, we need to cap the QM subsystem with a so-called link
atom (la in the figure). This link atom is present as a hydrogen atom in
the QM calculation step. It is not physically present in the MM
subsystem, but the forces on it, that are computed in the QM step, are
distributed over the two atoms of the bond. The bondlength itself is
constrained during the computations.
To make use of the QM/MM functionality in Gromacs, we have to
-
introduce link atoms at the QM/MM boundary;
- specify which atoms are to be treated at a QM
level;
-
specify the QM level, basis set, type of QM/MM interface
and so on.
These steps are a bit involved, and they require some editing in the
topology. Because we don't want to go too much into the technical
details here, we have prepared the topology, MD parameters file, and
initial structure. Please download the file and
unpack the tar-ball with
wget http://www3.mpibpc.mpg.de/groups/de_groot/compbio/p7/files/qmmm-system.tar.gz
tar xvzf qmmm-system.tar.gz
cd qmmm-system
Have a look into the structure file qmmm1.gro and into the topology
qmmm.top. You will find the link atoms with atom name LA. Note that
they are defined at so-called dummies (or "virtual sites") near the end of the
topology. That means
that they do not take part in the Newtonian dynamics - instead, their
positions are constructed at each step based on the positions of
nearby atoms.
Open the MD parameter file qmmm1.mdp, where the details
of the QM calculation are defined. Add the keyword AM1 for the option
QMmethod, and the basis STO-3G for option QMbasis. For now, the dimer
is neutral (set QMcharge to zero).
Question:
What is the spin multiplicity (QMmult) if we have fully occupied orbitals?
Let's start your first QM/MM simulation of 1ps.
For this part, we need a special mdrun binary that has been linked
with the MOPAC library. MOPAC
(Molecular Orbital PACkage) is a semiempirical quantum chemistry
program.
That mdrun and a suitable grompp been added to
the tar ball. Make the two binaries executable, load necessary libraries, and run the commands:
chmod +x grompp
chmod +x mdrun
export LD_LIBRARY_PATH=$(pwd)
./grompp -c qmmm1.gro -f qmmm1.mdp -n qmmm -p qmmm -o qmmm1.tpr
./mdrun -v -stepout 1 -deffnm qmmm1 -c qmmm1out.pdb
Since this run will take a while, we have provided the output files here. You may open a new terminal, download the
file, and unzip it and have a look at the trajectory with VMD:
wget http://www3.mpibpc.mpg.de/groups/de_groot/compbio2/p7/files/qmmmmd.tar.gz
tar xvzf qmmmmd.tar.gz
egrep -v ' Na | SOL' qmmm1out.pdb > qmmm1out_dna.pdb
vmd -e qmmm1.vmd
Question: Is the dimer stable, or do you observe any bond breaking
in the dimer?
Go back to Contents
The effect of electron uptake
 Now we want to study the effect of an electron uptake on the dimerized
thymine, as conducted in the presence of the enzyme Photolyase (see
introduction). Will the (unhealthy) bond break?
Now we want to study the effect of an electron uptake on the dimerized
thymine, as conducted in the presence of the enzyme Photolyase (see
introduction). Will the (unhealthy) bond break?
Open the mdp file qmmm2.mdp in an editor and set the QMcharge to -1,
corresponding to the state after an electron uptake. Now we have a
unpaired electron in the system.
Question: What is now the correct spin multiplicity (option QMmult)?
We start the
simulation from the last frame of the previous QM/MM simulation.
./grompp -f qmmm2.mdp -c qmmm1out.pdb -n qmmm -p qmmm -o qmmm2.tpr
./mdrun -v -stepout 1 -deffnm qmmm2 -c qmmm2out.gro
Since this run will again take a while, we have provided the output files here. You may again open a new terminal,
download the file, and unzip it and again, take a look at the
simulation in VMD:
wget http://www3.mpibpc.mpg.de/groups/de_groot/compbio/p7/files/qmmmmd2.tar.gz
tar xvzf qmmmmd2.tar.gz
vmd -e qmmm2.vmd
Question: Is the dimer stable, or does a bond break? Which bond is
breaking? Can you speculate why this and not the other bond breaks?
A final note: the first bond broke in less than a picosecond,
suggesting that this first rupture is essentially barrierless. If we
would simulate much longer, the second barrier would eventually break
as well - on a much longer time scale. In order to observe
the rupture, alternative simulations techniques are required such as
the calculation of free-energy profiles or conformational
flooding. These are, however, beyond the scope of this practical.
For questions or feedback please contact Bert de Groot / bgroot@gwdg.de