Setting up of a membrane protein simulation
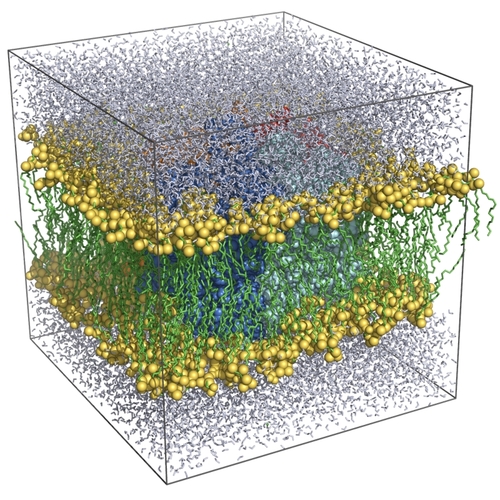
First of all, download the bovine aquaporin-1
structure (PDB code: 1J4N) currently available from the Protein Data Bank.
Have a look at the structure using pymol:
wget http://www3.mpibpc.mpg.de/groups/de_groot/compbio/p5/1J4N.pdb
pymol 1J4N.pdb
Before we can feed the structure to gromacs, we first have to delete
from the PDB file a detergent molecule that was co-crystallized with the
aquaporin. We do this with the unix 'grep' command. The '-v BNG' option tells
'grep' to print all lines except for the ones containing the string 'BNG':
grep -v BNG 1J4N.pdb > t.pdb
mv t.pdb 1J4N.pdb
now we can generate hydrogen positions and a gromacs topology using pdb2gmx:
gmx pdb2gmx -f 1J4N.pdb -o 1J4N_H.pdb
When asked for a force-field, select "15" (the OPLS/AA-L force-field), and
select the TIP4P water model.
Again examine the structure using pymol:
You may remember from the lecture that aquaporins are active as
tetramers, so if we want to study water permeation, we should simulate
an aquaporin tetramer. Since we only have the structure of one monomer
so far, we have to generate the coordinates of the other three
monomers based on symmetry. Usually, this can be done with software
like WHAT IF, but in this case it is more convenient to use a small
script that can be downloaded from our site. Before
we can use the script, we must make it executable:
wget http://www3.mpibpc.mpg.de/groups/de_groot/compbio/p5/maketet_i4.csh
chmod +x maketet_i4.csh
and now we can use it to generate the tetramer:
./maketet_i4.csh 1J4N_H.pdb tet.pdb
Have a quick view on the structure with pymol:
Now we'll merge the protein
system with a pre-equilibrated POPE membrane to yield the final simulation
system. Download the membrane from us, and have
a look at it with pymol:
wget http://www3.mpibpc.mpg.de/groups/de_groot/compbio/p5/pope.pdb
pymol pope.pdb
Before we can do the actual merge, we have to position the protein to
match the lipid coordinates:
gmx editconf -f tet.pdb -o conf.pdb -box 11.485 11.383 10.394
and now we can merge the two, removing all lipids and water molecules
that would overlap with the protein:
gmx solvate -cp conf.pdb -cs pope.pdb -o box.pdb
Analyse the result with pymol:
You now see the (almost) complete simulation system: a protonated
aquaporin-1 tetramer embedded in a solvated POPE lipid bilayer. Before
a production run can be started (to study water permeaion), the system
would first have to be energy-minimised, counter-ions would have to be
placed to compensate the net charge of the protein before a dynamical
equilibration can be started.
Question:
Aquaporins are passive water channels, driven by osmotic gradients across the
membrane. If we want to simulate such a process, how could we generate a
concentration gradient across the membrane? Wouldn't that dissipate across the
periodic boundaries of the simulation box rather than by permeation through
the channel?
Question:
Would it be possible to study permeation rates also without applying an
osmotic gradient? What could we learn from simulations in which water
molecules diffuse through the channel, without a driving force defined by the
osmotic gradient?
Since an actual simulation of water permeation would take too long for
this practical, we now make a leap in time, and pretend we have
already carried out the simulation.
Go back to Contents
Analysis of water permeation in aquaporin-1
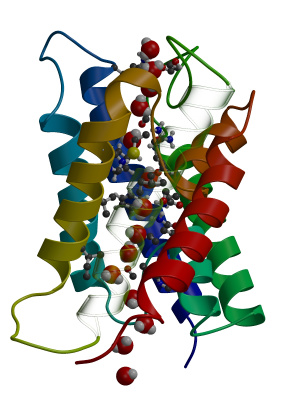 First of all, download the trajectory (fragment) full.xtc and a reference PDB file ref.pdb
and create a small animation:
First of all, download the trajectory (fragment) full.xtc and a reference PDB file ref.pdb
and create a small animation:
wget http://www3.mpibpc.mpg.de/groups/de_groot/compbio/p5/full.xtc
wget http://www3.mpibpc.mpg.de/groups/de_groot/compbio/p5/ref.pdb
gmx trjconv -s ref.pdb -f full.xtc -o movie.pdb -e 4100
Select "0" when prompted for a group.
Press the "play" button on the lower right of the main pymol button to
see the animation.
You will appreciate the difficulty of analysing
water permeation by simple visual inspection. This is why specialised
analysis tools have been developed to follow all water molecules
during the simulation, and to assess which water molecules participate
in permeation. Gromacs works with the concept of "index groups" which
are basically groups of atoms with a particular property. One could
for example define an index group containing all CA atoms of the
protein, and then use this index group to filter all CA atoms from an
MD trajectory for subsequent analysis. Such an index file has been
generated that contains a selection of water molecules that
participate in a particular permeation event. Download the index file and extract the protein plus a
selection of water molecules from the trajectory like this:
(First remove the old movie file, as it is quite big)
rm -f movie.pdb
wget http://www3.mpibpc.mpg.de/groups/de_groot/compbio/p5/index.ndx
gmx trjconv -s ref.pdb -f full.xtc -o movie.pdb -n
and select e.g. group 21.
Now view with pymol:
highlight one monomer of the aquaporin and the water molecules by typing (on the pymol prompt):
hide all
select aqua, res 1-227
show cartoon, aqua
select water, resn SOL
show spheres, water
Now highlight one of the water molecules by colouring it yellow:
select perm, res 3358
color yellow, perm
and press the "play button". You now see one water molecule that finds
its way from the extracellular face of the membrane to the cytoplasmic
side. Note that it might have gone just the opposite direction as
well, as no driving force (like an osmotic pressure) was applied
during the simulation. All permeation events, therefore, rest on pure
diffusion of water molecules through the aquaporin pores.
It is always important to test whether the simulation results agree
with experimental observations, whenever possible. In the aquaporin
case, we fortunately have an excellent opportunity to directly compare
our simulation results to experiments, namely to analyse whether the
permeation rate in the simulations is similar to the experimentally
determined one. In total, 16 full permeation events were observed
during the 10 ns simulation, in good agreement with the experimental
permeation rate.
Question:
Can you identify specific regions in the pore that you expect to be
particularly critical to make this channel a specific water channel?
Question:
Would you expect other small molecules like glycerol to pass through as well?
What about CO2?
Go back to Contents
Mechanism of proton exclusion
 It is critically important that no protons leak across the membrane, as
proton gradients are essential for bioenergetics, like for example in ATP synthesis.
Therefore, it is crucial that protons do not pass through aquaporins, alongside
with water molecules. This is a challenging task, as usually water, or water
filled pores, conduct protons efficiently. How can aquaporins, therefore,
accomplish the challenging task of being on the one hand an efficient water
pore, whereas on the other hand, actively block protons from passing?
It is critically important that no protons leak across the membrane, as
proton gradients are essential for bioenergetics, like for example in ATP synthesis.
Therefore, it is crucial that protons do not pass through aquaporins, alongside
with water molecules. This is a challenging task, as usually water, or water
filled pores, conduct protons efficiently. How can aquaporins, therefore,
accomplish the challenging task of being on the one hand an efficient water
pore, whereas on the other hand, actively block protons from passing?
Therefore, let us carefully check the behaviour of water molecules in the pore
to see if we can discover something about a possible mechanism of proton blocking.
Watch the last movie again and carefully check the behaviour of individual water molecules inside the pore
carefully.
Question:
Would you say that the water molecules are oriented randomly inside
the pore? Hint
Note that water molecules carry an electrical dipole:
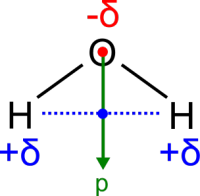
In an external electric field, water molecules will rotate to align with the
external field, to optimise their interaction with the field.
Question: Look again at the behaviour of
water molecules in the pore. How are the dipoles distributed? Could this be
due to an electric field inside the protein? How can the dipole behaviour
be explained in terms of interactions between the protein and passing
water molecules? How will this behaviour affect the permeation of
water? How will it affect the permeation of positively charged ions
and protons? What about negatively charged ions?
Have a close look at the distribution of amino acids in the pore. In pymol,
clicking on an atom gives you the residue number and type. Have a look at
this
table with amino acid types (best opened in a new window).
Question: Which amino acids do you think
are responsible for the effect observed above?
In addition to the interactions with the individual amino acids, note that
alpha helices carry a
macro dipole
, with the N-terminus carrying a positive charge and the C-terminus
carrying a negative charge.
Question: Have a close look at the
aquaporin structure again. In pymol, choose the "cartoons" representation. How
do you think the two short helices, that end in the middle of the pore, could
contribute to the electrostatic properties of the channel? Hint: look at the
location of the N-termini of these two helices.
Go back to Contents
Optional
Here we're going to have a go at protein design. Have another look at
this
table with amino acid types.
- Suggest two mutations (single amino acid substitutions) that in addition to water also conduct protons.
- Would it be possible, with one mutation, to change the channel such that
it conducts protons, but not water?
- In the proton conducting variants of aquaporin, which of these other
positively charged ions do you think might also be conducted? Sodium,
potassium, iron, calcium.
- Can you think of a mutation that instead of positively charged ion, would
conduct anions (negatively charged ions)?
Further references and advanced reading
- de Groot BL, Grubmüller H. The dynamics and energetics of water permeation and proton exclusion in aquaporins. Curr. Opin. Struct. Biol. 15: 176-183 (2005) [link]
- de Groot BL, Grubmüller H. Water permeation across biological membranes: Mechanism and dynamics of Aquaporin-1 and GlpF. Science. 294: 2353-2357 (2001) [link]
- Hub JS, de Groot BL. Mechanism of selectivity in aquaporins and aquaglyceroporins. Proc. Nat. Acad. Sci. 105: 1198-1203 (2008) [link]
For questions or feedback please contact Bert de Groot / bgroot@gwdg.de