Introduction

The last lecture gave an introduction into protein sequences
(or primary structures), and we have learnt which information can be
extracted on the sequence level. In short, these include:
- protein function prediction (when a homologue with known function is
annotated)
- secondary structure prediction (from the amino-acid propensities
in the sequence)
- tertiary structure prediction (when a homologue's structure is
known)
- identification of conserved (and possibly functionally or
structurally essential residues), by alignment with related sequences
- prediction of hereditary information, by alignment with related sequences
Today, we will focus on the last two aspects, as structural analyses
will follow later. First we will analyse the peptide hormone
insulin.
Go back to Contents
Multiple
sequence alignment of insulin
As you may know, insulin is essential for normal metabolism,
as it stimulates glucose uptake after a meal. Malfunction of insulin
leads to diabetes, which is characterized by decreased glucose
tolerance resulting from a relative deficiency of insulin (or,
alternatively, a lack of sensitivity to insulin on the receptor side).
For a sequence analysis of insulin, we obviously first need its sequence.
For this, we visit the SWISS-PROT database, which can be accessed via
the ExPASy Proteomics
Server.
- Using a browser on your PC, open the ExPASy web page in another browser window
(right click -> open in new window in mozilla), search for "UnipProtKB". ExPASy is a list of URL for tools you can use for bioinformatics.
- Click "Browse the ressource website" to go to UniProtKB
- On UniProt, search for "insulin".
- As you will see, there are hundreds of matches,but none directly corresponds to human insulin.
- This is because on the
sequence level, insulin is stored as a precursor. After synthesis, the
precursor protein is then split into the A and B chain that together
form the active form of insulin.
- Thus, we select the sequence of the
human insulin precursor : INS_HUMAN. Check the Uniprot ID, which should be P01308.
The SWISS-PROT entry of human insulin starts with some general
information on the sequence, starting with basic entry information,
the name and origin of the protein, literature references connected
with this sequence, and some comments concerning the function. This
section is followed by a number of cross-references to other databases
concerning insulin, like for example the Protein Data Bank (PDB),
where protein structures are stored. As we can see, lots of structural
information on insulin is available.
- In the section entitled
Feature viewer, we can learn how the precursor sequence is related to the
active form of the hormone.
- As can be seen in the "Molecule Processing" row of the graphic view, the first 24 residues are
a signal sequence, followed by a stretch of 30 residues (25-54) that
corresponds to the B chain of insulin, and residues 90-110 make up the
A chain of insulin.
- You can click on the rectangles to see their meaning.
- In the Variants row, you will find registered point mutations. Some of them are disease-causing, mostly various variants of diabetes.
Click on the dots for informations.
- Now go back to "Entry"
- On top of the page, just under the tabs ("Entry", "Feature viewer"), select "Dowload" and then "FASTA (Canonical)".
- A new tab opens with the sequence in the so-called "FASTA" format.
- Now we open a new browser window to run BLAST, which also can be accessed
from the ExPASy Proteomics
Server
- Search for "BLAST" and access the website UniProt BLAST
- Paste your sequence
- Run BLAST with the default parameters (Button on the bottom of the page)
- Wait for job completion
- After some time, we obtain the 250 database sequences closest to the human
insulin (precursor), sorted to their level of similarity.
- Select all sequences by ticking the checkmark to the left, in the table header near "Entry"
- Click "Add" on top of the table header (Shopping basket icon).
- Click the shopping basket icon on the very top of the page (shows a 99+ bubble)
- This opens our basket of selected sequence. Click Download from this Panel, then Download all (250), as format "Fasta (canonical)".
Be sure to check No for the "Compressed" setting.
- Save the file somewhere you will be able to find later
- Open a new browser window at the European Bioinformatics
Institute from which we will run the multiple alignment server.
Question: Using BLAST, the selected
sequences have already been aligned, to assess the similarity to our
target sequence. Why do we need to do another alignment?
- Search and access Clustal Omega
- In "Step 1", upload your sequence file that you got after running BLAST
- Run the job by clicking Submit on the bottom
- Click the Download Alignement File button and save the file to a known location.
We will now download and open an aligment viewer:
wget http://www.jalview.org/getdown/release/jalview-all-2.11.1.3-j1.8.jar
java -jar jalview-all-2.11.1.3-j1.8.jar
Close all windows within Jalview. Load the result of insulin by going to File > Input Alignment > From File and load the aligment file.
To focus on conserved residues, under "Colour", activate a coloring scheme, e.g. BLOSUM62 Score, and tick the "By Conservation" setting in the same menu.
You can keep the default conservation threshold.
This way,those residues that are highly conserved get highlighted according
to the selected threshold.
Question: Which are the most conserved
residues? Why might these residues be conserved? All cysteine (C) residues
seem highly conserved. What might be the reason?
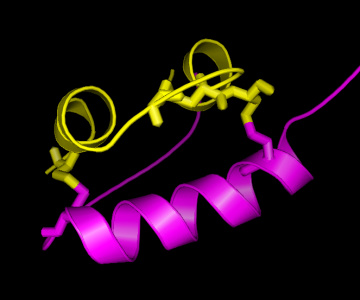
On the right, a picture of the insulin structure is shown, with the A
chain in yellow and the B chain in magenta. As can be seen, there are
two "bridges" connecting the A and the B chain, formed by Cysteine (C)
residues on both chains. This is an important structural feature of
insulin, strongly stabilising the structure. Therefore, it can be
easily understood that these C residues are among the strongly
conserved residues in the hormone. As is known from other structural
studies, residues interacting with the insulin receptor include:
the N-terminus of the A-chain (G-I-V-E), the C-terminus of the A-chain
(Y-C-N), and the C-terminus of the B-chain (G-F-F-Y), so also for
these residues there is a clear reason for their conservation. For the
other conserved residues, the reason for their conservation is less
clear, although their mutation has shown altered activity, hence
indicating a functional role.
Phylogenetic analysis of hemoglobin
Another application of multiple sequence analyses is the derivation of
evolutionary information, in particular the analysis of common ancestors
among different species, and their grouping (also known as taxonomy)
based on sequence similarity. This analysis is known as phylogenetic
analysis, and trees representing the sequence relationships are known
as phylogenetic trees.
 In this course we will generate two phylogenetic trees, and compare
the results, to see if the mutational pattern in the one protein
(and the associated phylogenetic tree) is similar to that of the
other. For this we will take the alpha and beta chain of hemoglobin.
Hemoglobin is the universal oxygen transporter in nature. It takes up
oxygens in the lungs (or gills for fish) and transports it via the
blood in red blood cells to the brain, muscle, or another destination
in the body where oxygen is required. In fact, the reason why blood is
coloured red is because of the hemoglobin. Hemoglobin contains iron,
which in that particular state is colored red, not unlike rust.
Although part of the same protein, the two sequences of the alpha and
beta chain have evolved
independently, and hence, two separate phylogenetic trees can be constructed.
In this course we will generate two phylogenetic trees, and compare
the results, to see if the mutational pattern in the one protein
(and the associated phylogenetic tree) is similar to that of the
other. For this we will take the alpha and beta chain of hemoglobin.
Hemoglobin is the universal oxygen transporter in nature. It takes up
oxygens in the lungs (or gills for fish) and transports it via the
blood in red blood cells to the brain, muscle, or another destination
in the body where oxygen is required. In fact, the reason why blood is
coloured red is because of the hemoglobin. Hemoglobin contains iron,
which in that particular state is colored red, not unlike rust.
Although part of the same protein, the two sequences of the alpha and
beta chain have evolved
independently, and hence, two separate phylogenetic trees can be constructed.
For the sequence retrieval, we follow the same procedure as we have
done above for insulin, first for the human hemoglobin alpha chain (search
for "human hemoglobin alpha"), and then for the hemoglobin beta chain.
- Go to UniProt, search for "human hemoglobin alpha"
- Open the page for alpha chain of hemoglobin (HBA_HUMAN - P69905) and the beta chain (HBB_HUMAN - P68871)
- On each page, click Blast (near the download link)
- In the settings, select "UniProtKB Swiss-Prot" as database (only high-quality, reviewed sequence entry)
- In the field "Restrict by taxonomy", enter "Eukaryota" (select in the autocomplete dropdown the entry with ID 2759)
- In Advanced parameters, Hits, select 50
- Submit jobs with these settings for both alpha and beta chains
- Once the jobs are completed, go to each of the BLAST result page
- Clear your sequence basket (top right) if it contains the previous results
- Using the "Identity" histogram on the left top, select the most similar sequences (say 92-100%), then deselect the epsilon and delta chain (name starting with HBE_ or HBD_)
- Add the selected sequence to your basket (Add button at the top of the table)
- Download the basket as FASTA (canonical), non-compressed. Name the file a recognizable name to distinguish the alpha and beta chains
- Take care not to mix the alpha and beta chains in your basket - you will need to clear the basket in between
- Go to ClustalW and upload the saved sequence (do this separately for alpha and beta chains)
- Download the alignment file for both chains
- Open JalView
- Open both alignment files
- From within each aligment window, in the Calculate window, select Calculate Tree. Leave parameters to defaults.
- The results are presented in a "cladogram", which enables easy comparison
of the individual groups. Click on the branches of the tree to invoke the color coding of the species belonging to the same branches.
- You can seach the sequence name (e.g. HBB_THEGE) in Uniprot if you do not know the latin name of the species
- Compare the tree you get for HBA and HBB
Questions:
- Which species are most similar to human?
- How related are humans to the Pygmy chimpanzee (PANPA), and the eurasian badge (MALME)?
- How does this example illustrate that it is dangerous to derive a
phylogenetic tree from the sequences of a single protein? hint
(due to
limited time, please go ahead with practical 4b and only then come back to
these optional exercises, if time allows).
Go back to  Protein function typically results from modular structural features called Domains. Due to evolutionary shuffling these domains can conservatively exist in proteins that share functional aspects.
Protein function typically results from modular structural features called Domains. Due to evolutionary shuffling these domains can conservatively exist in proteins that share functional aspects.