The major bottleneck of today's atomistic molecular dynamics (MD) simulations is that due to the enormous computational effort involved only processes at nanoseconds to microseconds time scales or faster can be studied directly. Unfortunately, apart from a few expections, relevant processes, such as chemical reactions or many large-scale conformational motions in biological macromolecules like proteins or DNA, occur at slower time scales and therefore are currently far out of reach for conventional MD. The flooding technique addresses this problem by inclusion of a flooding potential, Vfl, into the force field (see Figure). This flooding potential locally destabilizes the educt state and thereby significantly accelerates the escape from the initial energy well without affecting the reaction pathway. We have implemented the flooding method into the fast and freely available MD program Gromacs. (J. Comp. Chem. 2006,
27, 1693-1702. available online).
|
|
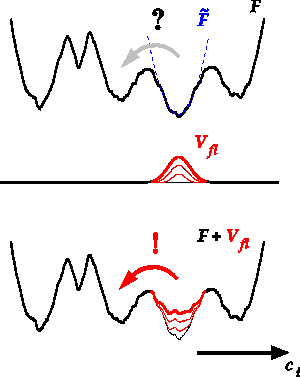
Fig. Principle of flooding. The free energy, F, along a collective coordinate, c, is approximated quasi-harmonically in the local educt minimum (blue dashed parabola). From this, a gaussian-shaped flooding potential Vfl (red) is constructed which destabilizes the initial well and accelerates the transition across the barrier (red arrow). |
|