
As described in the papers:
- Mars - robust automatic backbone assignment of proteins (2004) Young-Sang Jung and Markus Zweckstetter, J. Biomol. NMR, 30, 11-23.
- Backbone assignment of proteins with known structure using residual
dipolar couplings (2004) Young-Sang Jung and Markus Zweckstetter, J. Biomol. NMR, 30, 25-35.
- Automatic assignment of the intrinsically disordered protein tau with 441-residues (2010) Narayanan RL, Dürr UH, Bibow S, Biernat J, Mandelkow E, Zweckstetter M, J. Am. Chem. Soc., 132, 11906-11907.
Contact: mzwecks@gwdg.de
What is MARS ?
Download
Setup
Getting Started
Setting up input files
Setting up assignment parameters
How to run MARS
Output
Analysis of assignment / Testing
Directory structure
Important points to remember
For advanced users
What is MARS?
-
MARS is a powerful program for robust automatic backbone assignment of proteins. MARS is applicable to proteins with extreme chemical shift degeneracy such as high molecular weight proteins and partially disordered or fully intrinsically disordered proteins. MARS is successfully used in many labs world-wide.
- MARS works with a wide variety of NMR experiments, from 3D triple-resonance experiments up to the most complex 6D and 7D APSY experiments.
- MARS is interfaced with CCPNmr Analysis enabling direct easy access to automatic assignment without export or re-import of data.
- MARS results can be directly read into the program SPARKY. Thus, several cycles of automatic assignment and manual validation can be performed.
- MARS allows to fix sequential connectivity, residue-specific as well as residue-type assignments.
- MARS enables the use of 3D structures, through the calculation of residual dipolar couplings and scalar couplings, for improved resonance assignment.
- MARS is applicable to both deuterated and protonated proteins.
- MARS offers unsurpassed accuracy for proteins with incomplete data.
- MARS has excellent tolerance against erroneous chemical shifts.
- MARS combines secondary structure prediction with statistical chemical shift distributions, which are corrected for neighboring residue effects
MARS is interfaced with the program CCPNmr enabling direct easy access to automatic assignment without export or re-import of data.
MARS assignment results can also be read into the program SPARKY. This allows visual validation of the assignment results. Thus, several cycles of automatic assignment using MARS and manual validation on the screen can be performed, in order to complete assignment even in difficult cases.
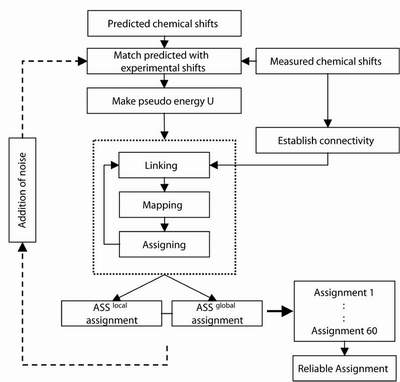
Key features
Download
- Obligatory
- parameter setup file (mars.inp)
- chemical shift table (SPARKY format)
- primary sequence (FASTA format)
- secondary structure prediction file (PSIPRED
format)
-
When a 3D structure is known and RDCs values are available
- PDB file
- RDC table (PALES
format)
- Optional
- table that allows restriction of the amino acid type and/or fixing of an assignment
- table that allows fixing of sequential connectivities between pseudoresidues.
- Prepare your chemical shift table.
- Get your primary sequence in FASTA format.
- Get a secondary structure prediction using the Psipred web server.
- Adjust the parameter setup file (mars.inp).
- Type 'runmars mars.inp'
- Assignment result filtered for high, medium and low reliablity
('assignment_AA.out').
- Assignment result including alternative assignments that show up with a 10 % probability ('assignment_AAs.out').
- The most likly assignment for each pseudoresidue ('assignment_PR.out').
- Summary of all possible connectivities ('connectivity.out').
- Summary of reduced possible connectivities ('connectivity_reduced.out').
- Chemical shift tables with updated assignments that can be read into
SPARKY ('sparky_all.out, sparky_CA.out, sparky_CA-1.out, sparky_CB.out, ...').
- Detailed information about predicted chemical shifts, number of reliable assignments, number of constraints for each pseudoresidue, matrices matching experimental and back-calculated chemical shifts and/or RDCs and pseudoenergy matrices at each iteration step ('mars.log').
- A Mars run is controlled by the parameter setup file (mars.inp).
This has to be adjusted to the available experimental data. Please
see below for a detailed description of the parameters.
-
Lines with a '#' sign as first character as well as empty line are
ignored.
Do not change the variable names such as nIter.mars.inp(MARSHOME/example/noStructure/1ubq/input/mars.inp)Optional parameters:-
fragSize: 5 # Maximum length of pseudoresidue fragments
cutoffCO: 0.25 # Connectivity cutoff (ppm) of CO [0.25]
cutoffCA: 0.2 # Connectivity cutoff (ppm) of CA [0.5]
cutoffCB: 0.5 # Connectivity cutoff (ppm) of CB [0.5]
cutoffHA: 0.25 # Connectivity cutoff (ppm) of HA [0.25]
cutoffHN: 0.02 # Connectivity cutoff (ppm) of HN [0.15]
cutoffN: 0.05 # Connectivity cutoff (ppm) of N [0.10]
cutoffnHN: 0.02 # Connectivity cutoff (ppm) of HN+1 [0.15]
cutoffnN: 0.05 # Connectivity cutoff (ppm) of N+1 [0.10]
fixConn: fix_con.tab # Table for fixing sequential connectivity
fixAss: fix_ass.tab # Table for fixing residue type and(or) assignment
pdb: 0 # 3D structure available [0/1]
resolution: NO # Resolution of 3D structure [Angstrom]
pdbName: NO # Name of PDB file (protons required!)
tensor: NO # Method for obtaining alignment tensor [0/1/2/3/4]
nIter: NO # Number of iterations [2/3/4]
dObsExh: NO # Name of RDC table for exhaustive SVD (PALES format)
dcTab: NO # Name of RDC table (PALES format)
deuterated: 0 # Protonated proteins [0]; perdeuterated proteins [1]
rand_coil: 0 # Folded proteins [0]; disordered proteins [1]
sequence: 1ubq_fasta.tab # Primary sequence (FASTA format)
secondary: 1ubq_psipred.tab # Secondary structure (PSIPRED format)
csTab: 1ubq_cs.tab # Chemical shift table
-
cyssTab: cys.tab # Table listing oxidized cysteines
csPred: 1 # Theoretical chemical shifts from MARS [1/0]
- chemical shift table
-
The chemical shift table follows the SPARKY
format. It consists of a header, pseudoresidues and chemical shifts.
The header has to be defined before the listing of chemical shift
values starts and includes the variable names for the chemical shifts.
Currently 10 different chemical shifts are supported and should
be indicated by 'Ca', 'Ca-1', 'Cb', 'Cb-1', 'CO', 'CO-1', 'HA',
'HA-1', 'H' and 'N'. These variable names have to be in the same
order as the columns for the different chemical shifts. The first
column has to be the pseudoresidue column and other columns are
chemical shift columns. Pseudoresidue means the name of the group
of peaks which share the same (or similar due to the experimental
imperfection) N and HN chemical shifts. Lines with a '#' sign
as first character as well as empty lines are ignored. Missing chemical
shift values have to be indicated by ' - '.
1ubq_cs.tab(MARSHOME/example/noStructure/1ubq/1ubq_cs.tab)Pseudoresidue names can consist of characters+number+characters and characters are optional. Pseudoresidues are distingushed by the whole set of characters. Therefore arbitrary names and numbers are allowed for pseudoresidues. (this is new in version 1.1.1)-
N CO-1 H CA-1 CA
PR_2 123.220 170.540 8.900 54.450 55.080
PR_3? 115.340 175.920 8.320 55.080 -
PR_4? 118.110 172.450 8.610 59.570 55.210
PR_5GLY 121.000 175.320 9.300 55.210 60.620
PR_6GLY 127.520 - 8.820 60.620 54.520
PR_7 115.400 177.140 8.730 54.520 60.470
PR_8 121.330 176.910 9.100 60.470 57.580
PR_9 105.590 178.800 7.630 57.580 61.400
PR_10?? 108.890 175.520 7.810 61.400 45.460
:
:
:
What to do if a chemical shift cannot unambigously attributed to a specific pseudoresidue?
MARS does not perform peak picking, referencing of spectra or grouping of peaks into pseudoresidues (PRs). In our lab we use SPARKY to perform these tasks. This allows visual control and refinement of pseudoresidues. When manually inspecting PRs, amide degeneracy can often be resolved, as peak shapes and the higher resolution in a 2D HSQC spectrum can be taken into account. If HN/N overlap remains, multiple spin systems should be provided to MARS comprising the full set of possible combinations of peaks. In order to avoid an unreasonable high number of PRs in these cases, ambiguous peaks can also be partially discarded, as MARS does not favor pseudoresidues with more complete chemical shift information during the assignment process. The suspicious peaks can be reinserted when running MARS a second or third time, after an initial MARS run was performed, the assignment results were visually validated using SPARKY and verified assignments were fixed.
Glycine: Fix the amino acid type in fix_ass.tab
MARS does not favor pseudoresidues with more complete chemical shift information. Therefore, it might happen that a spin system belonging to a glycine is wrongly assigned to another amino acid type, because both the connectivity might be fullfilled and the chemical shift matches sufficiently well. To avoid these problems, it is useful to fix the amino acid type of the spin systems that have uniquely been identified as glycines in the fix_ass.tab table.
- The primary sequence of the protein has to be in FASTA
format.
-
IMPORTANT: 'X' and 'Z' can not be
used for the characters of a sequence.
1ubq_fasta.tab (MARSHOME/example/noStructure/1ubq/1ubq_fasta.tab)-
> ubq
MQIFVKTLTGKTITLEVEPSDTIENVKAKIQDKEGIPPDQQRLIFAGKQLEDGRTLSDYN
IQKESTLHLVLRLRGG
- Secondary structure prediction table
-
This has to be in Psipred format. Use the Psipred
web server to get the table.
1ubq_psipred.tab (MARSHOME/example/noStructure/1ubq/1ubq_psipred.tab)-
PSIPRED PREDICTION RESULTS Key Conf: Confidence (0=low, 9=high)
Pred: Predicted secondary structure (H=helix, E=strand, C=coil)
AA: Target sequence Conf: 968896699888999867863189999999997689875658887777738887136726
Pred: CEEEEECCCCCEEEEEECCCCCHHHHHHHHHHHHCCCHHHEEEEECCEECCCCCCHHHHC
AA: MQIFVKTLTGKTITLEVEPSDTIENVKAKIQDKEGIPPDQQRLIFAGKQLEDGRTLSDYN
10 20 30 40 50 60 Conf: 8988889999950699 Pred: CCCCCEEEEEEECCCC
AA: IQKESTLHLVLRLRGG 70
If a 3D structure and experimental RDCs are available:
- PDB file
-
All standard PDB files
can be used (including MOLMOL
files).
IMPORTANT: When using shape-prediction all atoms in the PDB file will be used including pseudo atoms (ANI).
- Dipolar coupling input
Experimental dipolar couplings are supplied according to the PALES table format:
- The protein sequence should be given as shown by one or more "DATA SEQUENCE" lines. Space characters in the sequence will be ignored.
- The table must include columns for residue ID, three-character
residue name and the atom name for both atoms that are involved
in the dipolar coupling as well as the dipolar coupling itself,
its error and a weighting factor. Segment ID and Chain ID
are optional.
IMPROTANT: The atom notation must match that of the PDB file. - The table must include a "VARS" line that labels the corresponding columns of the table.
- The table must include a "FORMAT" line that defines the data type of the corresponding columns of the table.
- Lines with a '#' sign as first character as well as empty lines are ignored.
-
Example dipolar coupling table (excerpts):
DATA SEQUENCE MQIFVKTLTG KTITLEVEPS DTIENVKAKI QDKEGIPPDQ QRLIFAGKQL DATA SEQUENCE EDGRTLSDYN IQKESTLHLV LRLRGG VARS RESID_I RESNAME_I ATOMNAME_I RESID_J RESNAME_J ATOMNAME_J D DD W FORMAT %5d %6s %6s %5d %6s %6s %9.3f %9.3f %.2f 2 GLN N 2 GLN HN -15.524 1.000 1.00 3 ILE N 3 ILE HN 10.521 1.000 1.00 4 PHE N 4 PHE HN 9.648 1.000 1.00 5 VAL N 5 VAL HN 6.082 1.000 1.00 1 MET C 2 GLN HN 3.993 0.333 3.00 2 GLN C 3 ILE HN -5.646 0.333 3.00 3 ILE C 4 PHE HN 1.041 0.333 3.00 4 PHE C 5 VAL HN 0.835 0.333 3.00 1 MET C 2 GLN N 2.651 0.125 8.00 2 GLN C 3 ILE N -3.768 0.125 8.00 3 ILE C 4 PHE N 1.463 0.125 8.00 4 PHE C 5 VAL N -1.726 0.125 8.00 2 GLN N 2 GLN HN -15.524 1.000 1.00 3 ILE N 3 ILE HN 10.521 1.000 1.00 4 PHE N 4 PHE HN 9.648 1.000 1.00 5 VAL N 5 VAL HN 6.082 1.000 1.00 1 MET HA 1 MET CA -38.341 1.000 0.50 2 GLN HA 2 GLN CA 11.662 1.000 0.50 3 ILE HA 3 ILE CA 18.424 1.000 0.50 4 PHE HA 4 PHE CA 26.733 1.000 0.50 - The chemical shift of oxidized cysteines differs signifcantly from that of reduced ones. Therefore, the residue number of cysteines involved in a disulphide bond should be listed in a file (i.e. residues 3,23,36 and 58 are involved in a disulphide bond). Reduced cysteines should not be listed here.
cys.tab (MARSHOME/example/noStructure/1ubq/cys.tab)
-
3
23
36
58
- The theoretical chemical shifts used by MARS for matching fragments to the primary sequence can be modified manually. This allows the usage of experimental chemical shifts from a homologues protein or using an improved chemical shift prediction software. User-supplied 'theoretical' chemical shifts can be included by: (i) Setting the csPred parameter in mars.inp to '1'. (ii) Run Mars to obtain a table with theoretical chemical shifts predicted by MARS (Cs_pred.tab). (iii) Modify Cs_pred.tab. (iv) Change the csPred parameter from '1' to '0' in mars.inp. (v) Run Mars again.
- When additional information such as specific amino acid type
labeling or initial manual assignments are available assignment
of pseudoresidues can be restricted to single or to a set of residues.
The first column has to be a pseudoresidue name followed by residue
numbers or amino acid types to which the assignment should be
restricted. Assignments can be fixed one by one by specipying
the corresponding residue numbers or restrict it to a whole residue
fragment by specifying the starting and ending residue number
(inclusive) connected by '-' (without a blank in between the start
and end number!). At the same time, amino acid types can be fixed
by specifying the corresponding one letter code. More than one
amino acid type can be specified by concatenation of the corresponding
one letter codes (i.e. attach additional one-letter codes without
blank in between).
fix_ass.tab (MARSHOME/example/noStructure/1ubq/fix_ass.tab)
-
PR_3 3
PR_10 10-15 23 34
PR_12 12 34-36
PR_13 13
PR_14 14 16 HKT
PR_15 LFR 66-69 13-16 9 71
PR_16 EVA
- Also sequential connectivities can be fixed. This is especially
useful when assignment is done iteratively by Mars and manually.
The first and second column are pseudoresidue names. The first
column is the name of the pseudoresidue for which the intra-residual
chemical shift can be connected to the inter-residual chemical
shift of the pesudoresidue in the second column.
fix_con.tab (MARSHOME/example/noStructure/1ubq/fix_con.tab)
-
PR_2 PR_3 PR_3 PR_4 PR_4 PR_5 PR_11 PR_12
PR_12 PR_13 PR_13 PR_14 PR_25 PR_26 PR_26 PR_27
- cutoffCO is the tolerance value (ppm) for matching intra- and inter-residual chemical shifts of C'.
- cutoffCA is the tolerance value (ppm) for matching intra- and inter-residual chemical shifts of Ca.
- cutoffCB is the tolerance value (ppm) for matching intra- and inter-residual chemical shifts of Cb.
- cutoffHA is the tolerance value (ppm) for matching intra- and inter-residual chemical shifts of Ha.
- The first column is the residue number of the protein; the second
column is the pseudoresidue that the residue is assigned to. The third
column indicates the degree of reliability of each assignment. Three
levels of reliability are distinguished:
H indicates high reliablity as defined in the MARS paper. M and L do not fulfill all the criteria required for H reliablity and the specific criteria employed are adjusted automatically according to the completeness of the input data. Please see below for the robustness of assignments labelled as M and L. F indicates assignments fixed by the user.assignment_AA.out
-
MET_1
GLN_2 PR_2 (M)
ILE_3 PR_3 (M)
PHE_4 PR_4 (H)
VAL_5 PR_5 (H)
LYS_6
THR_7
LEU_8
THR_9 PR_9 (M)
GLY_10 PR_10 (H)
LYS_11 PR_11 (H)
THR_12 PR_12 (H)
ILE_13 PR_13 (H)
THR_14 PR_14 (H)
LEU_15 PR_15 (H)
GLU_16 PR_16 (M)
VAL_17
GLU_18
PRO_19
SER_20 PR_20 (L)
ASP_21
THR_22
:
:
:
- The first column is the residue number of the protein. Additional
colums list pseudoresidues that can be assigned to this resdiue. Numbers
in parenthesis are assignment probablities. Only pseudoresidues with
an assignmnent probablity of higher than 10% are shown. assignment_AA.out
is a subset of the assignments here.
assignment_AAs.out
-
MET_1
GLN_2 PR_2 (96)
ILE_3 PR_3 (100)
PHE_4 PR_4 (100)
VAL_5 PR_5 (100)
LYS_6 PR_6 (63) PR_8 (30)
THR_7 PR_7 (76)
LEU_8 PR_8 (61)
THR_9 PR_9 (100)
GLY_10 PR_10 (100)
LYS_11 PR_11 (100)
THR_12 PR_12 (100)
ILE_13 PR_13 (100)
THR_14 PR_14 (100)
LEU_15 PR_15 (100)
GLU_16 PR_16 (100)
VAL_17 PR_17 (73)
GLU_18
PRO_19
SER_20 PR_20 (86)
ASP_21 PR_21 (65)
THR_22 PR_57 (33)
:
:
:
- assignment_PR.out lists the most likely assignment for each
pseudoresidue present in the input chemical shift table. The first
column is the pseudoresidue and the second is the residue (to which
the pseudoresidue can be assigned to most likely). NOTE: 'The
most likely assignment' does not mean reliable assignment and
two pseudoresidues can also be assigned to one residue. The information
present in assignment_PR.out is useful if a pseudoresidue is
not assigned to any residue in assignment_AAs.out and one asks
himself what it might be assigned to.
assignment_PR.out
-
PR_2 GLN_2
PR_3 ILE_3
PR_4 PHE_4
PR_5 VAL_5
PR_6 LYS_6
PR_7 THR_7
PR_8 LEU_8
PR_9 THR_9
PR_10 GLY_10
PR_11 LYS_11
PR_12 THR_12
PR_13 ILE_13
PR_14 THR_14
PR_15 LEU_15
PR_16 GLU_16
PR_17 VAL_17
PR_18 GLU_18
PR_20 GLN_40
PR_21 GLN_41
PR_22 SER_57
:
:
:
- In connectivity.out all possible sequential connectivities
between pseudoresdiues are listed. All numbers are pseudoresidue numbers.
The first column (closed by '-->') is the pseudoresidue number for
which connectivities are listed. If no additional entries are present
no connectivities could be found for that pseudoresidue. Otherwise,
all pseudoresidue numbers are listed for which the inter-residual
chemical shift can be matched to the intra-residual chemical shift
of the pseudoresidue in the first column.
connectivity.out
-
PR_2 --> PR_3 PR_5 PR_35 PR_43 PR_69 PR_74
PR_3 --> PR_4 PR_23 PR_30 PR_56
PR_4 --> PR_3 PR_5 PR_29 PR_35 PR_43
PR_5 --> PR_6 PR_8 PR_71
PR_6 --> PR_2 PR_7 PR_49 PR_55
PR_7 --> PR_6 PR_8
PR_8 --> PR_9 PR_21
PR_9 --> PR_10
PR_10 --> PR_11 PR_48 PR_76
PR_11 --> PR_12
PR_12 --> PR_13
PR_13 --> PR_14
PR_14 --> PR_15
PR_15 --> PR_16 PR_44
PR_16 --> PR_17 PR_69 PR_74
PR_17 --> PR_18 PR_60
PR_18 --> PR_16
PR_20 --> PR_9 PR_21
PR_21 --> PR_22 PR_40 PR_41 PR_50 PR_73
PR_22 --> PR_23 PR_56
- Detailed information about assignment parameters, percentage of
expected intra- and inter-residual Ca, Cb, Co and Ha chemical shifts
present in the input chemical shift table, predicted chemical shifts,
number of reliable assignments, number of constraints for each pseudoresidue,
matrices matching experimental and back-calculated chemical shifts
and/or RDCs and pseudoenergy matrices at each iteration step.
mars.log
-
------------------------------------------------------------------------------------------
fragSize: 5 # Maximum length of pseudoresidue fragments
cutoffCO: 0.25 # Connectivity cutoff (ppm) of CO [0.25]
cutoffCA: 0.2 # Connectivity cutoff (ppm) of CA [0.5]
cutoffCB: 0.5 # Connectivity cutoff (ppm) of CB [0.5]
cutoffHA: 0.25 # Connectivity cutoff (ppm) of HA [0.25]
fixConn: fix_con.tab # Table for fixing sequential connectivity
fixAss: fix_ass.tab # Table for fixing residue type and(or) assignment
pdb: 0 # 3D structure available [0/1]
resolution: NO # Resolution of 3D structure [Angstrom]
pdbName: NO # Name of PDB file (protons required!)
tensor: NO # Method for obtaining alignment tensor [0/1/2/3/4]
nIter: NO # Number of iterations [2/3/4]
dObsExh: NO # Name of RDC table for exhaustive SVD (PALES format)
dcTab: NO # Name of RDC table (PALES format)
deuterated: 0 # Protonated proteins [0]; perdeuterated proteins [1]
sequence: 1ubq_fasta.tab # Primary sequence (FASTA format)
secondary: 1ubq_psipred.tab # Secondary structure (PSIPRED format)
csTab: 1ubq_cs.tab # Chemical shift table
------------------------------------------------------------------------------------------
# of AA: 76
# of PRO: 3
# of GLY: 6
# of Assignable AA: 72
# of PR: 72
CA: 100.0 CB: 0.0 CO: 0.0 HA: 0.0
Ca: 100.0 Cb: 0.0 Co: 100.0 Ha: 0.0
AC RC RW
--------------------
53 11 0
--------------------
55 13 0
54 13 0
--------------------
54 20 0
53 20 0
--------------------
:
:
:
:
:
- Check the connectivity.out to verify that your chemical
shift table has been made properly. If there are problems in your
chemical shfit table due to miscalibration of spectra and(or) many
pseudoresidues grouped incorrectly, you will see many missing sequential
connectivities in the connectivity.out table.
- MARS keeps reliablility of assignment even for highly degenerate
and/or incomplete data sets and labels assignments according to
three different levels of reliability, H (high reliable assignment),
M (medium reliable assignment), L (low reliable assignment).
The following table shows the robustness of Mars assignment for
different proteins, different connectivity cutoffs and ranked according
to H, M and L reliability. From this table
it becomes clear that even medium reliable assignments are basically
error-free and only for less stringent connectivity cutoffs assignments
labelled as L contain a few errors.
-
Table 1. (BMRB chemical shifts used
for the test. / COcutoff: 0.25, CAcutoff: 0.5, CBcutoff: 0.5)
Tested proteins Used chemical shfits # of AA (assignable AA) H (Correct/Incorrect) M (Correct/Incorrect) L (Correct/Incorrect) Maltose binding protein C', Ca, Cb 723 (654) 639/ 0 9/ 0 0/ 0 Maltose binding protein Ca, Cb 370 (335) 303/ 0 18/ 1 0/ 0 EIN of the phospoenolpyruvate Ca, Cb 259 (248) 232/ 0 6/ 0 0/ 0 E-cardherin domains II Ca, Cb 227 (167) 76/ 1 5/ 1 12/ 9 Human prion protein Ca, Cb 210 (190) 130/ 0 9/ 0 5/ 0 Superoxide dismutase C', Ca, Cb 192 (117) 101/ 0 2/ 1 2/ 0 Calmodulin/M13 complex C', Ca 148 (144) 37/ 0 7/ 0 17/ 2 E. coli EmrE C', Ca, Cb 110 (74) 35/ 0 6/ 0 10/ 3 Human ubiquitin Ca, Cb 76 (72) 72/ 0 0/ 0 0/ 0
Table 2. (BMRB chemical shifts used for the test. / COcutoff: 0.15, CAcutoff: 0.2, CBcutoff: 0.4)Tested proteins Used chemical shfits # of AA (assignable AA) H (Correct/Incorrect) M (Correct/Incorrect) L (Correct/Incorrect) Maltose binding protein C', Ca, Cb 723 (654) 639/ 0 9/ 0 0/ 0 Maltose binding protein Ca, Cb 370 (335) 324/ 0 7/ 0 2/ 0 EIN of the phospoenolpyruvate Ca, Cb 259 (248) 246/ 0 0/ 0 0/ 0 E-cardherin domains II Ca, Cb 227 (167) 102/ 0 7/ 0 19/ 0 Human prion protein Ca, Cb 210 (190) 127/ 0 4/ 0 5/ 0 Superoxide dismutase C', Ca, Cb 192 (117) 104/ 0 0/ 0 1/ 0 Calmodulin/M13 complex C', Ca 148 (144) 142/ 0 0/ 0 0/ 0 E. coli EmrE C', Ca, Cb 110 (74) 58/ 0 4/ 0 0/ 0 Human ubiquitin Ca, Cb 76 (72) 72/ 0 0/ 0 0/ 0
Table 3. (Raw peak lists used for the test. / CAcutoff: 0.3, CBcutoff: 0.5, HAcutoff: 0.05)Tested proteins Used chemical shfits # of AA (assignable AA) H (Correct/Incorrect) M (Correct/Incorrect) L (Correct/Incorrect) Z domain protein Ca, Cb, Ha 71 (67) 65/ 0 0/ 0 0/ 0 Z domain protein Ca, Cb 71 (67) 34/ 0 6/ 0 2/ 0
- Connectivity_reduced.out filters all detected connectivities
reported in connectivity.out according to high reliable assignments.
This can be useful for manual refinement of a Mars assignment.
- Spectra calibration and proper peak grouping are the most important
points.
- When grouping inter- and intra-chemical shifts try to use the
same spectrum for extraction of inter- and intra-chemical shfits
of a given atom type.
For example, when you want to get intra- and interresidual chemical shifts of Ca, extract both chemical shifts from the HNCA spectrum. Only take the interresidual Ca chemical shift from a one way connectivity spectrum like HN(Co)CA, if the interresidual peak in the HNCA is too weak or overlapping.
In that case, bigger connectivity cutoffs (cutoffCO, cutoffCA, cutoffCB, and cutoffHA) have to be used due to imperfections in spectrum calibration.
Nevertheless, Mars does not care where you got the intra- and inter-chemical shifts from!
- Be careful of folded peaks!
The MARS software is freely available for academic users.
Industry and commercial users are invited to obtain a license at MARS Commercial license.
MARS (version 1.2)
-
The download provides a compressed tar archive with a MARS
executable and example files. The archive can be unpacked
with a command like the following:
tar -xvzf mars-1.2_linux.tar.gz
Graphical user interface for MARS (version 1.0)
-
There is also a simple graphical user interface for MARS available:
PALES (home)
For assignment using residual dipolar couplings the software PALES has to be installed in addition. This is only required if a 3D structure is known.Setup
-
Open your .cshrc files in your home directory. Add the three
lines below to your .cshrc file. The setenv LC_NUMERIC
command is only required in case you have a German Unix version.
setenv MARSHOME directoryName
alias runmars directoryName/runmars
setenv LC_NUMERIC "C"
setenv PALESHOME directoryName
(directoryName is the name of the directory that contains
the binary and script files.)
An example:
setenv MARSHOME /home/spine/local/MARS/mars-1.0_linux/bin
alias runmars $MARSHOME/runmars
setenv LC_NUMERIC "C"
The MARS GUI may be started by executing one of the following files from a X11 window:
marsgui-1.0-linux (for LINUX)
marsgui-1.0-MAC or marsgui-1.0-MAC-AQUA (for MAC)
Getting started
Input
MARS is a program for backbone assignment of 13C/15N labeled proteins. Accordingly, following input is required:
How to run Mars
Output
Setting up input files
Obligatory
Optional
Setting up assignment parameters
fragSize:
-
Sequential connectivity is established by matching inter- and intra-residual
chemical shifts. Fragments comprising up to fragSize pseudoresidues
are searched for exhaustively. The maximum segment length fragSize
is a compromise between the desired total execution time of a MARS
assignment run and the ability to reliably place PR segments onto
the protein sequence.
According to our tests a fragSize of 5 is large enough to get reliable assignments (pseudoresidue fragments with length five can in most cases be placed uniquely into the protein sequence when intra- and inter-residual Ca and Cb chemical shifts are available).
For smaller proteins or if more computing power is available larger fragment sizes (six or seven) can be employed. This is expected to be useful if, for example, no Cb chemical shift information is available.
cutoff
Cutoff values should be determined according to the resolution of the spectra. If chemical shifts were obtained from standard HNCACB, CBCACONH and HNCO experiments reasonable values will be
Ex.) cutoffCO: 0.1 cutoffCA: 0.5 cutoffCB: 0.5 cutoffHA: 0.1
fixConn
-
This is optional. If you want to fix
sequential connectivities, prepare a table like fix_con.tab
and specify the table name, otherwise set the fixConn parameter
to NO.
NOTE: At one iteration step MARS generates 60 assignment solutions and extracts reliable assginments from these solutions. After the first iteration step MARS automatically fixes reliable assignments and reliable sequential connectivities obatined from previous iteration steps without user intervention. The iteration is continued until the number of reliable assignments does not increase any more. Therefore, one can see fixed assignments and fixed sequential connectivities on the screen during a MARS run although the user didn't fix anything at the start of MARS.
Ex.)
fixConn: NO or fixConn: fix_conn.tab
deuterated
-
If a protein is perdeuterated, set the deuterated parameter
to 1. Otherwise put it to 0.
Ex.)
deuterated: 0 or deuterated: 1
sequence
-
Specifiy the name of the file that contains the primary sequence of
your protein in FASTA format.
Ex.)
sequence: 1ubq_fasta.tab
secondary
-
Specifiy the name of the file that contains the secondary structure
information of your protein in PsiPred format.
Ex.)
secondary: 1ubq_psipred.tab
csTab
-
Specifiy the name of the file that contains the experimental chemical
shifts (SPARKY format).
Ex.)
csTab: 1ubq_cs.tab
If no 3D structure or RDCs are available,
put the additional paramters as below:
pdb: 0 resolution: NO pdbName: NO tensor: NO nIter: NO dObsExh: NO dcTab: NO
If a 3D structure and experimental RDC are
available,
following parameters have to be set up:
pdb
-
Put the pdb flag pdb to 1,
in order to use RDCs and the known 3D structure (otherwise set it
to 0 ).
Ex.)
pdb: 1
resolution
-
Specify the resolution of your crystal structure. If you don't konw
the resolution of the structure because it is a homology model, set
the resolution to ~ 4.0.
In this case it will be useful to perform multiple assignment runs
with decreasing values for the resolution parameter (suggested range
is 2.0 < resolution < 6.0).
The optimum value corresponds to the assignment run where the maximum
number of reliable assignments was obtained.
Ex.)
resolution: 1.8
pdbName
-
Name of file containing the coordinates of the 3D structure. All standard
PDB files (including Molmol)
can be used. IMPORTANT: Protons have to be present.
Ex.) pdbName: 1ubq.pdb
tensor
-
Method for obtaining an initial estimate of the alignment tensor.
Four different modes are available that can automatically be accessed
by specifying 1, 2, 3 or
4. The standard mode is
3.
-
If 1 is selected, MARS
will use a gridSearch for estimating the orientation
of the alignment tensor.
If 2 is selected, MARS will use exhaustive back-calculation (exhSVD). (dObsExh parameter has to be setup!)
If 3 is selected, MARS will use singular value decomposition (SVD).
If 4 is selected, MARS will use shape-prediction (shapePred).
Ex.)(It is recommended to use 1 or 3 for the tensor parameter. Modes 2 and 4 require additional knowledge or RDCs in nearly neutral alignment media.)
tensor: 3
nIter
-
MARS refines the initial alignment tensor estimate (obtained by the
tensor method specified above) several times using SVD based
on the reliable assignments obtained in previous iteration steps.
Here, the number of refinement steps of the alignment tensor, nIter,
can be defined. According to our tests 2
refinement steps are enough.
Ex.)
nIter: 2
dObsExh
-
For exhaustive back-calculation (tensor mode 2)
an RDC table is required that contains RDCs of a specific amino acid
type. If the tensor mode is 1,
3 or 4, put the
dObsExh parameter to NO.
Ex.)
dObsExh: NO or dObsExh: dObs_1ubq_GLY.tab
dcTab
-
Name of file that contains the experimental RDC values (in PALES format).
Ex.)
dcTab: dObs_1ubq.tab
Output
Analysis of assignment
Directory structure
MARSHOME:
$MARSHOME/bin $MARSHOME/example
$MARSHOME/example/noStructure $MARSHOME/example/Structure
$MARSHOME/example/noStructure/1ubq $MARSHOME/example/noStructure/1zym $MARSHOME/example/noStructure/1dmb
$MARSHOME/example/Structure/1ubq $MARSHOME/example/Structure/1zym $MARSHOME/example/Structure/1dmb
Important points to remember
For advanced users
Tensor
-
Grid search
-
1116 alignment tensor orientations are systematically sampled and
for each orientation the deviation D(i,j) between experimental and
back-calculated RDCs is determined (equation 1).These 1116 orientations
are obtained in a two-step procedure. First, the z-axis of the molecule
samples uniformly 122 points on a unit sphere that were determined
by a double cubic lattice method (Eisenhaber et al., 1995). Only
one quarter of these points have to be taken into account due to
the inversion symmetry of RDCs. In a second step, the molecule is
rotated around the z-axis in steps of 10? giving a highly uniform
and efficient sampling of all possible tensor orientations. All
sampled orientations are ranked according to their corresponding
D(i,j) values (equation 1) and the lowest D(i,j) value indicates
the best estimate for the experimental alignment tensor. Thus, both
chemical shifts and RDCs are used, making the extraction of the
tensor orientation more robust.
-
In this method resonances belonging to a specific amino acid type
are identified and all possible assignments are searched exhaustively
for best-fit of back-calculated to experimental RDCs. The permutation
that shows the best agreement identifies the experimental alignment
tensor. exhSVD can be applied as long as at least one amino acid
type is less than eleven times present in the primary sequence of
the protein.
-
When the assignment is known, an alignment tensor can be obtained
by best-fitting experimental dipolar couplings to a 3D structure
using singular value decomposition (SVD) (Losonczi et al., 1999).
Thus, a two-stage strategy for structure-enhanced assignment can
be devised. In the first stage, RDCs are not used and partial assignments
are obtained using only chemical shift matching or a combination
of chemical shift matching and sequential connectivity information,
i.e. using a common backbone assignment strategy. This assignment
is assumed to be correct and a alignment tensor is obtained from
best-fitting experimental RDCs to the 3D structure. The accuracy
of the alignment tensor will depend on the percentage of correct
assignments that were obtained in the first assignment phase. Tests
show that, even when the percentage of correct assignment is below
50%, the alignment tensor is very close to its correct orientation.
This is due to the fact that wrong assignments are not randomly
distributed on the primary sequence of the protein. As Ca/Cb chemical
shifts depend very much on the type of secondary structure, exchange
of assignments mainly takes place between residues located on the
same type of regular secondary structure. In addition, b-strands
are often part of sheet structures, i.e. different strands are quite
collinear. Therefore, residues located in these strands have similar
RDCs and back-calculated alignment tensors are not severely affected
by an assignment where pseudoresidues are interchanged between two
b-strands (data not shown). The applicability of this approach depends
mainly on the amount and type of chemical shift information available,
i.e. how good is the assignment without RDCs.
-
Second, molecular alignment tensors of proteins dissolved in nearly
neutral bicelles can be predicted from the three-dimensional shape
of the protein (Zweckstetter and Bax, 2000). This is particularly
attractive as the only information necessary is the 3D structure,
i.e. RDCs can be used in the same way as chemical shifts for structure-enhanced
assignment. A disadvantage is that this method requires measurement
of dipolar couplings in a nearly neutral alignment medium restricting
the general applicability of RDC-enhanced assignment. However, recent
results indicate that prediction of alignment tensors from 3D structures
can be extended to a wide variety of alignment media, when electrostatic
interactions are taken into account (Zweckstetter et al., 2003).
Thus, this promises to be a valuable approach for obtaining approximate
alignment tensors before the start of RDC-enhanced assignment.
What is MARS ?
Download
Setup
Getting Started
Setting up input files
Setting up assignment parameters
How to run MARS
Output
Analysis of assignment / Testing
Directory structure
Important points to remember
For advanced users
Lab home-page at DZNE
Home-page DZNE
Home-page MPIBPC
last updated 2014-11-10