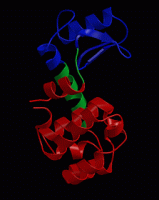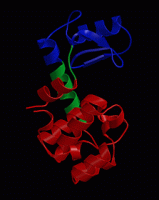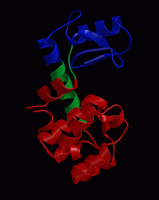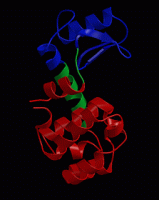
wget http://www3.mpibpc.mpg.de/groups/de_groot/compbio1/p4/148L.pdb pymol 148L.pdb
select sub,(resn NAG,MUB) color red,sub show sticks,sub
select cat,(resi 11,20,26) color yellow,cat show sticks,cat
dss show cartoon
Since it would take too long to calculate a long enough MD simulation during the course, we have prepared a simulation of the (substrate-free) enzyme. Now download this trajectory fragment and view it in pymol:
wget http://www3.mpibpc.mpg.de/groups/de_groot/compbio1/p4/t4l.pdb pymol t4l.pdb
Question: Which motions can you identify? How can you imagine those motions to be crucial for the protein's function?
wget http://www3.mpibpc.mpg.de/groups/de_groot/compbio1/p4/ref.pdb wget http://www3.mpibpc.mpg.de/groups/de_groot/compbio1/p4/md1_backbone.xtc
gmx trjconv -s ref.pdb -f md1_backbone.xtc -o md1.pdb -e 1000
pymol md1.pdb
gmx covar -s ref.pdb -f md1_backbone.xtc
ls -lrt
xmgrace eigenval.xvg
gmx covar -s ref.pdb -f md1_backbone.xtc -o largest_eigenval.xvg -last 30
xmgrace largest_eigenval.xvg
gmx anaeig -s ref.pdb -f md1_backbone.xtc -filt filter1.pdb -first 1 -last 1 -skip 100
pymol filter1.pdb
As you can see, the animation is kind of jerky. This is because the even such large-scale motions do not occur smoothly, but stochastically. To see a smooth animation of the motion along the first eigenvector, we can artificially interpolate between the extreme conformations sampled during the simulation along this eigenvector:
gmx anaeig -s ref.pdb -f md1_backbone.xtc -extr extreme1.pdb -first 1 -last 1 -nframes 30
pymol extreme1.pdb
Question: How would you describe the motion along the first and second eigenvector? What happens with the substrate-binding cleft in these two motions?
To see the contrast with the other eigenvectors, now repeat the above procedure for eigenvector 15 (or 20, or any other number of your choice).
Question: The backbone of T4 lysozyme contains 486 atoms (162 residues and 3 backbone atoms per residue). How many eigenvectors/values would you expect? How many eigenvalues should be exactly zero, and why? How many should be approximately zero?
For a more quantitatve analysis, we can project the trajectory onto individual eigenvectors, and display the projections as a function of time:
gmx anaeig -s ref.pdb -f md1_backbone.xtc -proj -first 1 -last 6 xmgrace -nxy proj.xvg
Another way to visualise the sampled conformations in the subspace spanned by the eigenvectors is a so-called two-dimensional projection:
gmx anaeig -s ref.pdb -f md1_backbone.xtc -2d -first 1 -last 2 xmgrace 2dproj.xvg
We can use a PCA not only to analyse an MD simulation and extract the main concerted motions from a trajectory, but also for a comparison of multiple simulations or for the comparison to an ensemble of experimental structures, in terms of dominant, collective motions. For T4 lysozyme, a large number of x-ray structures has been determined (if you search the PDB for "T4 lysozyme", you'll find more than 400 structures). We have collected 38 non-redundant x-ray structures of T4 lysozyme, which are all together collected in allpdb_bb.xtc. Just like on the simulated trajectory, we can perform a PCA on the collection of experimental structures:
wget http://www3.mpibpc.mpg.de/groups/de_groot/compbio1/p4/allpdb_bb.xtc gmx covar -s ref.pdb -f allpdb_bb.xtc -nopbc
gmx anaeig -s ref.pdb -f allpdb_bb.xtc -extr extreme1_xray.pdb -first 1 -last 1 -nframes 30 gmx anaeig -s ref.pdb -f allpdb_bb.xtc -extr extreme2_xray.pdb -first 2 -last 2 -nframes 30
Question: What kind of motion is represented by the first and second eigenvector? How does this compare to the results from the simulation?
For a quantitative comparison of the simulation and the experimental strutures, we'll project both the experimental structures and the simulated structures onto the eigenvectors extracted from the x-ray structures:
gmx anaeig -s ref.pdb -f md1_backbone.xtc -2d md_2d.xvg -first 1 -last 2 gmx anaeig -s ref.pdb -f allpdb_bb.xtc -2d xray_2d.xvg -first 1 -last 2 xmgrace md_2d.xvg xray_2d.xvg
Question: How do the x-ray and MD ensemble compare to each other? Why do you think the MD simulation does not sample the x-ray configurations on the extreme left of the plot (the most open x-ray conformations)? And why do you think the twisting mode is sampled with a larger amplitude in the simulation than in the ensemble of x-ray structures?
Two additional simulations are available for T4 lysozyme, that started from different x-ray structures. Download the two backbone trajectories to your local disk: md2_backbone.xtc and md3_backbone.xtc. Cross project also these two trajectories onto the x-ray eigenvectors and visualise all three simulations and the ensemble of x-ray structures projected onto the first two eigenvectors of the x-ray ensemble.
wget http://www3.mpibpc.mpg.de/groups/de_groot/compbio1/p4/md2_backbone.xtc wget http://www3.mpibpc.mpg.de/groups/de_groot/compbio1/p4/md3_backbone.xtc gmx anaeig -s ref.pdb -f md2_backbone.xtc -2d md_2_2d.xvg -first 1 -last 2 gmx anaeig -s ref.pdb -f md3_backbone.xtc -2d md_3_2d.xvg -first 1 -last 2 xmgrace md_2d.xvg xray_2d.xvg md_2_2d.xvg md_3_2d.xvg
Question: Which of the three simulations covers most of the crystallographic structures? Do you consider the simulations long enough to sample the most relevant conformations of the protein?
Question: All three simulations were of the substrate-free protein. Would you expect a more 'open' or a more 'closed' state as the most probable configuration in this situation?
Question:
Alternatively, we could have carried out a Normal Modes analysis of this
protein, approximating the energy landscape by a multidimensional harmonic
energy minimum. Does the motion along the first PCA eigenvectors appear
(quasi)-harmonic? If yes, which force-constant would you estimate?
gmx anaeig -s ref.pdb -f md1_backbone.xtc -proj -first 1 -last 1 xmgrace proj.xvg
Question: For Argon in the gas phase (without periodic boundary conditions), what kind of results would you expect from a PCA analysis? Particularly, what would you expect the eigenvalue spectrum to look like?
Carry out a short simulation of Argon in the gas phase, using this topology, these starting coordinates, and these MD parameters. Now carry out a PCA over the Argon trajectory:
wget http://www3.mpibpc.mpg.de/groups/de_groot/compbio1/p4/topol.top wget http://www3.mpibpc.mpg.de/groups/de_groot/compbio1/p4/argon_gas.pdb wget http://www3.mpibpc.mpg.de/groups/de_groot/compbio1/p4/md.mdp gmx grompp -f md.mdp -c argon_gas.pdb gmx mdrun -v echo 0| gmx trjconv -f traj_comp.xtc -s topol.tpr -o nojump.xtc -pbc nojump echo 0| gmx covar -s argon_gas.pdb -f nojump.xtc -nofit -nopbc
Question: Is the eigenvalue spectrum as expected?
Have a look at the projections onto the principal eigenvectors with:
gmx anaeig -s argon_gas.pdb -f nojump.xtc -proj -first 1 -last 6
xmgrace proj.xvg
Question:
Do you note the similarity of these projections to cosines? What implications
does this have for protein trajectories, where the principal mode projections
appear as cosines? answer.