Introduction

During part a) of the practical the emphasis was on protein sequence
retrieval and analysis. We will now slowly turn towards protein
structure and focus on what can be deduced on a protein's structure
based on it's sequence. Specifically, we will predict the structure of
a small protein based on its sequence similarity to another protein,
with known structure.
We are going to predict the structure of the alpha-dendrotoxin from
the green mamba snake. This is the toxin contained in the venom of the
green mamba that endangers the prey after a bite.
First, we will extract the toxin sequence from the UniProt
database. Open a browser, search for "alpha-dendrotoxin" in UniProt after selecting "UniProtKB" in the dropdown to the left of the search field. Click on the required sequence
(it should be the first one listed in the UniProtKB database: VKTHA_DENAN (P00980)), and on the
the top of page, click Dowload, and save the FASTA (Canonical) file to a local file.
Go back to Contents
Secondary structure prediction of alpha-dendrotoxin
As discussed in the lectures, a protein's sequence (primary structure)
can be used as a basis for a prediction of its secondary
structure. The principle of such methods is based on the fact that
different amino acids and amino acid combinations have different
preferences for different types of secondary structure. Alanine, for
example is often found in alpha helices, whereas prolines are known to
destabilise helices. Automated procedures exist that have optimised
prediction algorithms against a databank of proteins with known
structures. One such prediction program is available as an online server:
the
JPred4 Secondary Structure Prediction server.
- Open the FASTA file, and copy the sequence (second line with capital one-letter code only) into the input sequence field.
- Submit the job ("Make prediction").
- If prompted about match found in the PDB, choose "Continue" to use the prediction software regardless of the available structure
- Wait a bit until the result page appears
The prediction is presented in the line "jnetpred". A continuous line (-) stands for unstructured
(i.e. neither helix nor sheet), an arrow stands for extended, or sheet, and a red cylinder
stands for helix. The "JNETCONF" bar graph indicates the confidence of the system in its prediction (higher is better).
As you can see, the server predicts the protein to
start from the N-terminus with an unstructured loop, followed by two
beta strands and a short helix.
Question:
Where is the model less sure of its prediction? Why?
Go back to Contents
Structure prediction of tertiary structure
We have the sequence of our protein of interest, we need a
suitable template structure of a homologous protein on the basis of
which we can build a model of the venom structure. For this, we visit
the Protein Data Bank. The
protein we're going to use as a template is the bovine (cow)
pancreatic trypsin inhibitor. In the
search field, search for "trypsin inhibitor bovine". Among the search
results select "4PTI" (or search for it directly).
Alternative, download the structure from our site and have a look at it by typing:
wget http://www3.mpibpc.mpg.de/groups/de_groot/compbio2/p14/4PTI.pdb
pymol 4PTI.pdb
Please note that the commands in the gray boxes can be easily transferred to the command prompt with
copy-and-paste (select text by dragging the mouse over it with the left mouse
button pressed, and paste by pressing the middle mouse button).
We are going to build a model using an
internet server, the SWISS-MODEL
server. Paste the sequence of the snake venom in the sequence window (or
use the SWISS-PROT access code: P00980) and upload 4PTI.pdb via "Add Template File". Now, submit the request by hitting the "Build Model" button. Depending on the load of the
server, it may take a couple of minutes for the model to finish. Once the calculation has finished, go to "Models", click "Model 01" and download the stucture in PDB format. Save the structure as swissmodel.pdb. You are going to need the correct reference structure (1DTX.pdb) to compare against as well. In case the calculation takes too long, we also provide the coordinates. Download them with:
wget http://www3.mpibpc.mpg.de/groups/de_groot/compbio2/p14/swissmodel.pdb
wget http://www3.mpibpc.mpg.de/groups/de_groot/compbio2/p14/1DTX.pdb
Assuming your model is called "swissmodel.pdb", superimpose the structures in pymol:
pymol 1DTX.pdb swissmodel.pdb
and in pymol, align the two molecules by clicking on the A in the right panel on the line 1DTX. In the menu, go to "align", "All to this (*/Ca)".
Question:
How similar/different are the model and the reference structure?
Go back to Contents
Structure prediction: AlphaFold
Homology modeling has been recently supplanted, with great media attention, by the Deep Learning-based AlphaFold in structure prediction
contest CASP. While AlphaFold is not
available as a webserver, and requires considerable computational ressources to run locally, we can access its prediction for most genetic sequences
found in bioinformatics databases like UniProt.
Go the the AlphaFold Structure Database and search for the UniProt accession number of the toxin (P00980).
Download the PDB file as "toxin-alphafold.pdb".
We will then compare the SwissModel model and the AlphaFold model, as well as the experimental structure.
pymol 1DTX.pdb fit_swiss.pdb toxin-alphafold.pdb
Align the AlphaFold structure in PyMol by clicking on the A in the right pane on the line toxin-alphafold. In the menu, go to "align", "All to this (*/Ca)".
You can now click the names in the right pane to show/hide the three versions and judge which model looks closest to the experimental structure.
You may want to look at the turn between the two beta sheets.
To briefly sketch a picture of how AlphaFold works, let's look at this diagram from the
AlphaFold Nature Paper:
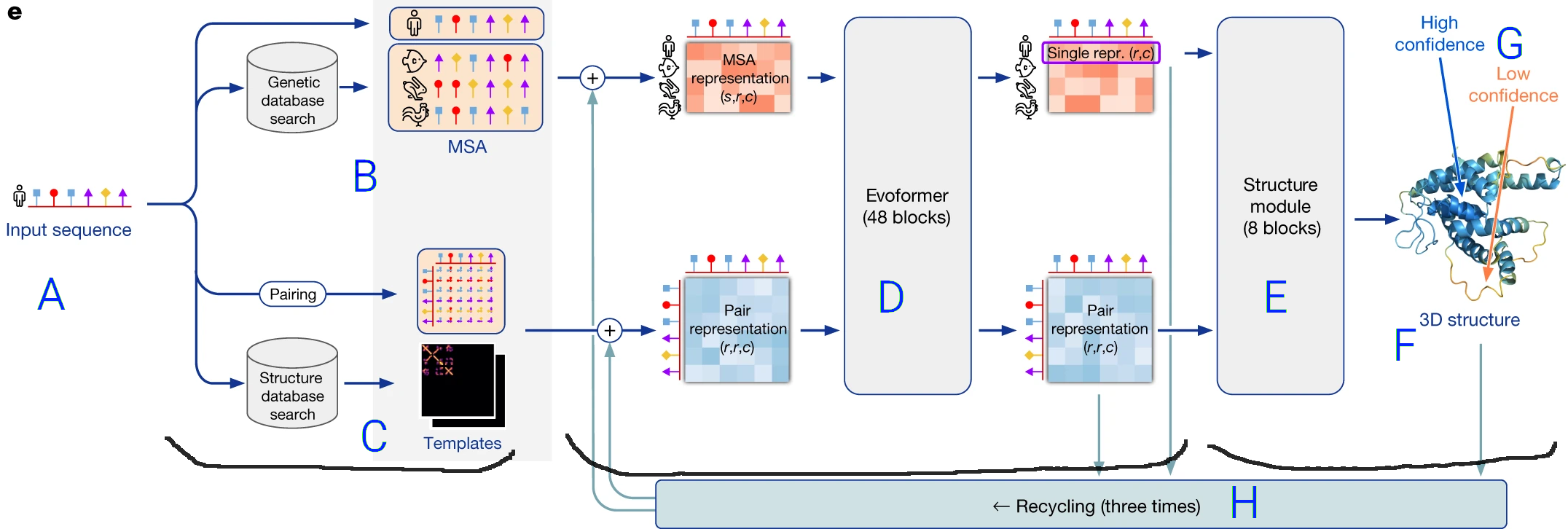
- The prediction pipeline starts with the protein sequence (amino acids in order) (A).
- It performs two operations shown as path in the flow diagram:
- On top, a multiple sequence alignement (MSA) (B), in which similar protein sequences are sought (in a BLAST-like process).
This MSA allows identification of conserved residues and correlated mutations in the sequence, which are translated to a format understandable
by a neural network ("MSA representation").
- On the bottom, the structure of proteins with similar sequences is retrieved from a structure database (C),
and these templates
are also converted to a format understable by the neural network ("Pair Representation").
- Both the
MSA and the template structures are used as inputs to a Transformer neural network, named the Evoformer (D),
which is a specific network architecture that efficiently transforms one kind of information into another exploiting
common patterns and spasity: here sequence and related structure informations are transformed into an internal representation
of the structure of the protein
of interest.
- Finally, the output of the Transformer is fed to a last neural network (E) that translates
the internal representation into an actual structure (F),
as well as a confidence score self-judging the accuracy of the modelling for each part of the protein (G).
- The final structure is refined by an iterative process (H) where it is taken as a template for another round of neural network
prediction.
This describes the process needed to predict stucture. But the neural network required training to be as accurate as it is. This process requires
the use of training examples, which are sequence with a known-good structure. For AlphaFold, the training examples are structure from the Protein Data Bank.
In an automated,
iterative and highly computationally intensive process, the sequences were fed to the pipeline, and the predicted structures were compared
to the PDB structures. The settings ("weights") of the neural network were adjusted until the prediction was similar enough to the reference structure.
This means that, in addition to the structure database (C), the neural networks
(D) and (E) also contain distilled information from the Protein Data Bank
Question:
Which of the predicted structures (SwissModel or AlphaFold) seems closer to the experimental structure?
Question:
Given the above description of how AlphaFold works, is this a fair comparison to SwissModel?
Further references
- Protein Structure Prediction Center [link]
- W. Priovano and J. Heringa, Protein Secondary Structure Prediction Methods in Molecular Biology V. 609, Part 3, P 327-348 (2010). [link]
- J. Moult, A decade of CASP: progress, bottlenecks and prognosis in protein structure prediction Curr. Opin. Struct. Biol. 15:285-289 (2005). [link]
Go back to Contents