
This practical provides an introduction to coarse-grained (CG) modeling. A CG model reduces the number of degrees of freedom in a system by reducing the number of interaction sites, resulting in a model that is computationally less expensive. In this practical, we will optimize the bonded terms of the coarse-grained force field with the help of all-atom (AA) molecular dynamics (MD) simulation. The practical builds on concepts covered in previous lectures, such as the potential of mean force (PMF) and free energy calculation. We will carry out the Martini (coarse-grained model) parametrization of a dodecane molecule using target atomistic simulation of a dodecane solution. The goal of the optimization process is to match a mapped atomistic distribution to a coarse-grained one.
To start the optimization of a coarse-grained force field, we need to generate reference data first. To begin the practical, download the relevant files and unpack them using the following commands:
wget http://www3.mpibpc.mpg.de/groups/de_groot/compbio/p_coarse_grained/files/CGPractical.tar.gz tar xzvf CGPractical.tar.gz cd CGPractical/AA
First have a look at our AA system.
pymol start.gro
To load a trajectory type in pymol
load_traj traj_aa.xtc
To get a perspective on the speed-up in CG simulation compared to AA, we will first run an AA MD simulation and then compare it to the CG simulation later in the practical. Start the simulation and terminate it after some time using Ctrl+C.(Write down the speed (ns/day))
gmx grompp -f md.mdp -p topol.top -c start.gro -o aa.tpr gmx mdrun -v -deffnm aa
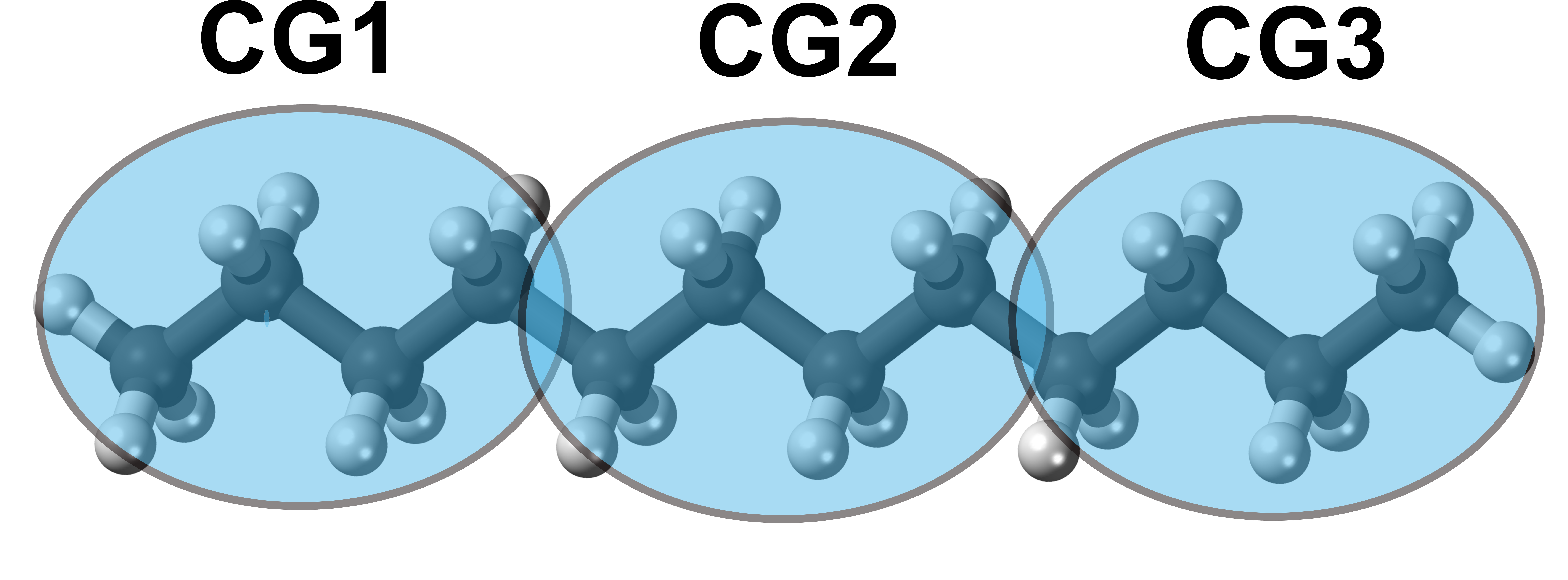
Now that we have our reference data, we need to decide on the resolution of the CG model and the mapping. Mapping defines which atoms are assigned to which CG beads. The Martini model generally assumes a 4:1 mapping, which means 4 heavy atoms are assigned to 1 CG bead. We will follow the same mapping strategy and map 4 carbon atoms with their attached hydrogen atoms to 1 CG bead.
With the defined mapping, we can proceed to transform the AA trajectory into CG simply by calculating the center of mass for each group of atoms within a CG bead. To transform our trajectory, we first need to generate an index file where the index of every atom is assigned to a CG bead. Since we have 201 molecules in our system, we will use a script to write 603 index groups (3 CG beads per molecule).
python mkindex.py
To see which atom index is assigned to which CG bead, open this Python script with any text editor.
You should now have the index file, cg_ind.ndx. To use it with gmx traj, request 603 groups (number of CG beads in the system), set the -com flag, and provide the PBC-treated AA trajectory, the compiled topology, and the new index file:
seq 0 602 | gmx traj -f traj_aa.xtc -s aa.tpr -oxt traj_cg.xtc -n cg_ind.ndx -com -ng 603 seq 0 602 | gmx traj -f traj_aa.xtc -s aa.tpr -oxt cg.gro -n cg_ind.ndx -com -ng 603 -e 0
Now we have our mapped trajectory at hand. Take a look at the mapping.
pymol cg.gro start.gro
To make the CG representation more visible, type the following in pymol:
show spheres, cg set sphere_scale, 0.5, cg
With your mapped trajectory file ready, you can now use gmx distance and gmx angle to extract bonded distributions of the beads. To do this, you'll need to generate index files that specify bonds and angles. Since generating hundreds of atom pairs and triplets will require some scripting, use mkbonded_ind.py to generate index files.
python mkbonded_ind.py
Now you should have index files bonds.ndx and angles.ndx containing all bonds and angles in your system. With these, we can proceed to calculate distributions.
gmx distance -f traj_cg.xtc -len 0.47 -tol 0.3 -n bonds.ndx -oh bond_dist.xvg gmx angle -n angles.ndx -f traj_cg.xtc -od angle_dist.xvg
Select the bonds in the index file "0" and press Ctrl+D when generating the bond distribution.
Take a look at both distributions
xmgrace bond_dist.xvg xmgrace angle_dist.xvg
To obtain bonded parameters for our CG model, we will use the Boltzmann inversion method. In this method, the effective bond and angle potentials between CG sites are derived from bond and angle distributions. To estimate the spring constants for bonds and angles, we will assume that our atomistic distributions are Gaussian.
Question:
If we assume that the probability distribution is Gaussian, how does the PMF formula simplify? What is the spring constant?
Run a following scripts for every distribution to get parameters of a harmonic potential.
python getparams.py bond_dist.xvg python getparams.py angle_dist.xvg
With these parameters, we can now create a topology for the CG representation of dodecane. Change directory and replace "?" in ddec_cg.itp file with appropriate parameters. We also need a coordinate file for the CG simulation. We can use the one we already generated.
cd ../CG cp ../AA/cg.gro . gedit ddec_cg.itp
Now run minimization and equilibration of the system. In the first round of simulation, atom types in the coordinate and topology files do not match. To ignore the warning, add the -maxwarn 1 flag.
gmx grompp -f min.mdp -p topol.top -c cg.gro -o min.tpr -maxwarn 1 gmx mdrun -v -deffnm min gmx grompp -f eq.mdp -p topol.top -c min.gro -o eq.tpr gmx mdrun -v -deffnm eq
Now, with the equilibrated system, we can finally run the CG simulation and compare it to the AA simulation
gmx grompp -f md.mdp -p topol.top -c eq.gro -o md.tpr gmx mdrun -v -deffnm md
Question:
By how much is CG simulation faster than AA? Is the speed-up linear with the number of coarse-grained atoms (On average, we replaced 13 atoms with 1 CG bead)?
Visualizing the CG system is a little bit tricky compared to AA because bond lengths in the CG system are generally much larger than in any AA bond, which many software do not recognize. To visualize our system, run the following commands
gmx trjconv -f md.xtc -o cg.pdb -pbc mol -conect yes -s md.tpr -e 0 gmx trjconv -f md.xtc -o traj_cg.xtc -pbc mol -s md.tpr
Press "0" to select the whole system.
Now you can open cg.pdb file with pymol. To load trajectory use load_traj command in pymol
Compute bond and angle distributions for our CG system and compare them to the mapped AA distributions using xmgrace.
cp ../AA/*.ndx . gmx distance -f traj_cg.xtc -len 0.47 -tol 0.3 -n bonds.ndx -oh bond_dist.xvg gmx angle -n angles.ndx -f traj_cg.xtc -od angle_dist.xvg
xmgrace bond_dist.xvg ../AA/bond_dist.xvg -legend load xmgrace angle_dist.xvg ../AA/angle_dist.xvg -legend load
Question:
How do these distributions compare? Is this a good agreement? Why do we see a discrepancy between AA and CG distributions?
In a real optimization process of the CG force field, parameters of the force field are iteratively updated until the CG distribution matches the AA distribution within some tolerance. Here we can update our parameters manually to improve agreement between distributions. Replace angle in ddec_cg.itp file with 180 degree and set force.c in range between 30-40 kj/mol/rad^2 and rerun CG simulaiton. Compare all 3 distribution
gmx grompp -f md.mdp -p topol.top -c eq.gro -o md.tpr gmx mdrun -v -deffnm md gmx trjconv -f md.xtc -o traj_cg.xtc -pbc mol -s md.tpr gmx angle -n angles.ndx -f traj_cg.xtc -od angle_dist_new.xvg xmgrace angle_dist.xvg ../AA/angle_dist.xvg angle_dist_new.xvg -legend load
With the optimized CG force field, we can now compare our system's fundamental statistical mechanical quantities, e.g., the radial distribution function (RDF).
To compute and compare the RDF, use the following commands:
gmx rdf -f traj_cg.xtc -excl -s md.tpr -o rdf_cg.xvg gmx rdf -f ../AA/traj_cg.xtc -excl -s md.tpr -o rdf_aa.xvg xmgrace rdf* -legend load
For both commands, specify 2 selections to calculate RDF, both as '0', so we calculate the RDF of every CG bead with every other CG bead.
Additionally, we can set RDF as a target for optimizing the non-bonded potential (taken from Martini here).
Question:
How do these RDFs compare? Based on this distribution, do you think that additional refinement is needed for non-bonded interactions?
gmx msd -f traj_cg.xtc -o msd_cg.xvg -selrpos res_com -s md.tpr gmx msd -f ../AA/traj_cg.xtc -o msd_aa.xvg -selrpos res_com -s md.tpr xmgrace msd* -legend load
Question:
In which systm diffusion is faster? Why do you think diffusion is faster? How does this modifies effective time in CG system?
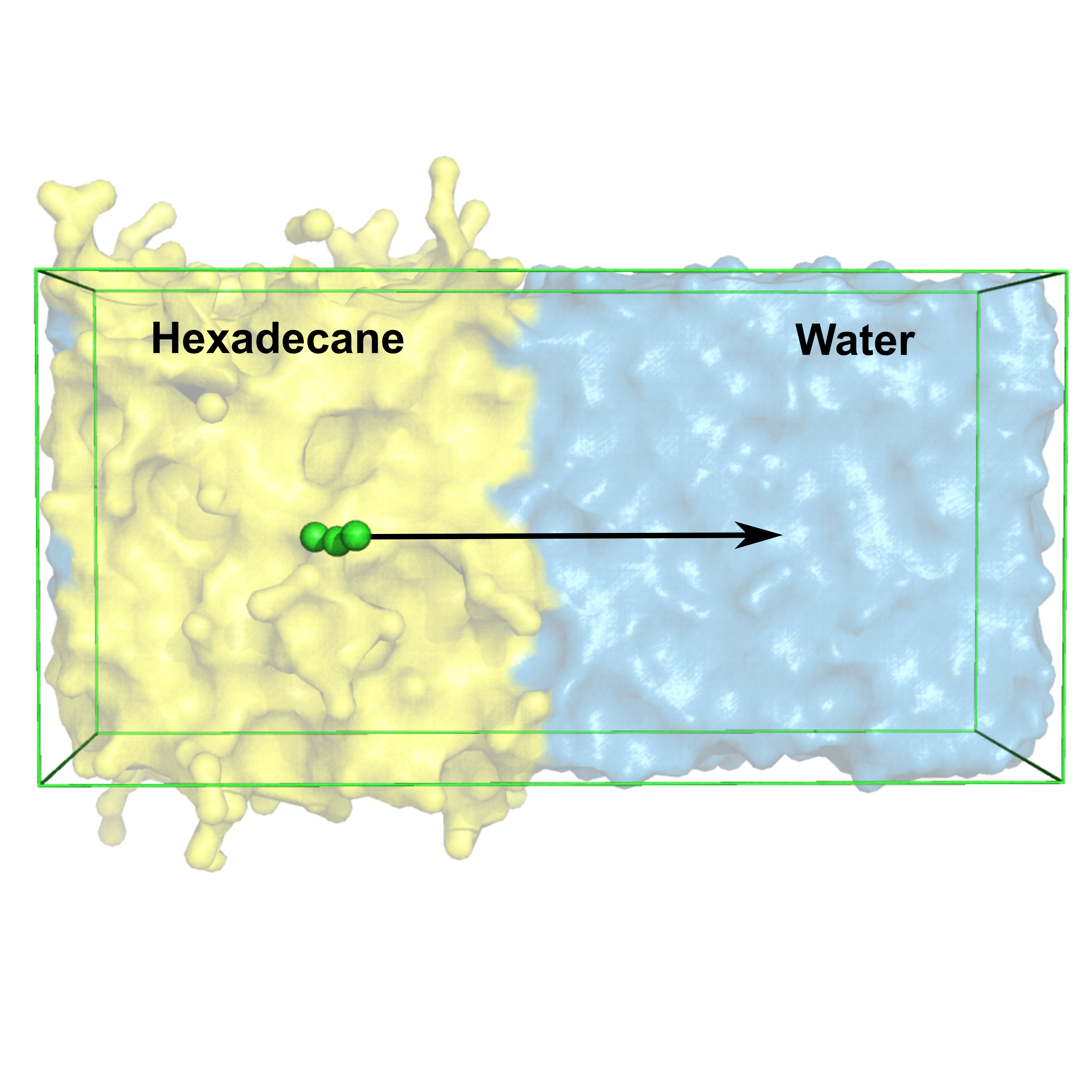
cd ../US gmx wham -it tpr_files.dat -if pullf_files.dat -o -hist -unit kT
Plot resulting profile profile.xvg with xmgrace