Examples for using tCONCOORD:
A simple tCONCOORD simulation of a protein (Staphylococcal nuclease)
Download the pdb file 1ey4.pdbRemove water:
$ grep -v HOH 1ey4.pdb > tmp.pdbUse gromacs pdb2gmx to add hydrogens and prepare energy minimization
$ pdb2gmx -f tmp.pdb -o gmx.pdb -ignh -ff oplsaaDownload .mdp file for energy minimization em.mdp
Do energy minimization with gromacs
$ grompp -f em.mdp -c gmx.pdb $ mdrun -c em.pdbDownload tCONCOORD parameter file for normal runs: input.cpf
Do constraint definition with tdist:
$ tdist -s em.pdbGenerate 100 structures with tdisco:
$ tdisco -n 100
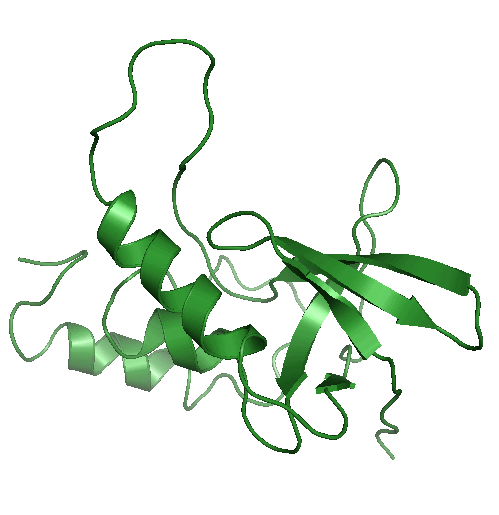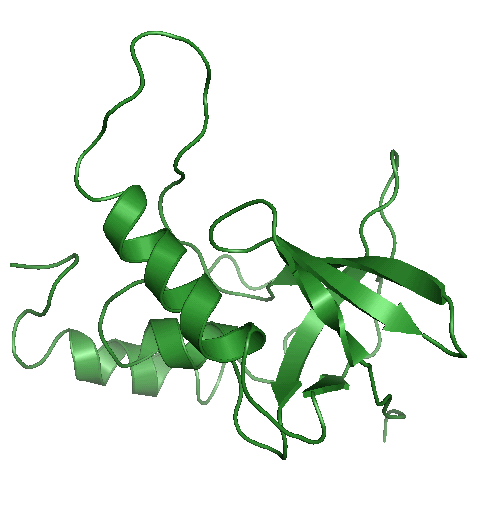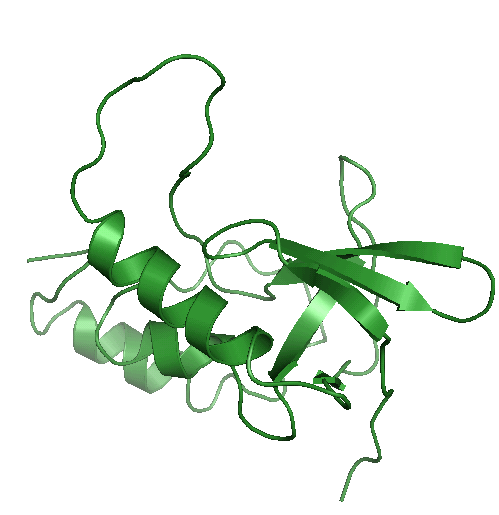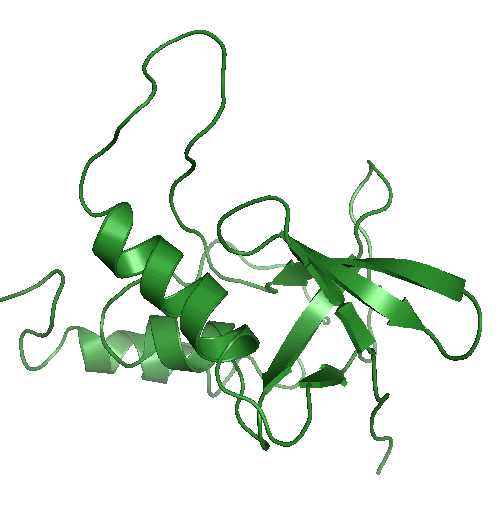
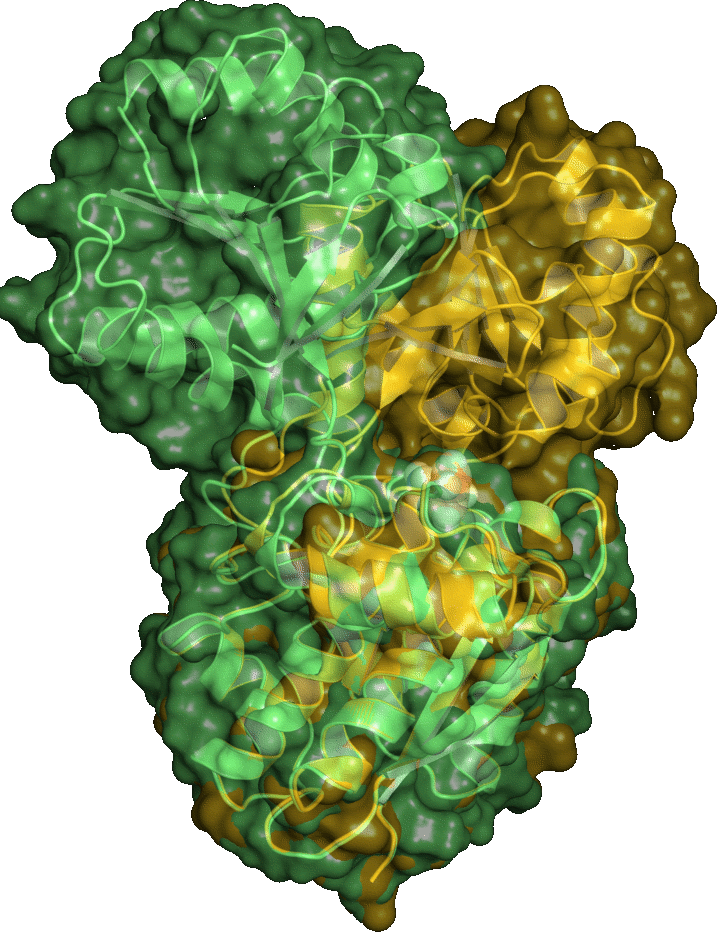
Using position constraints / Generating alternate loop configurations
Here we generate a set of alternate loop conformations for a protein structure. Download the minimized structure 1ugu_min.pdbWe take the residues 49-65 as loop and generate an index file which contains two groups, the loop and the rest.
$ make_ndx -f 1ugu_min.pdb > r 49-65 ( this is now group 10) > !10 >qNow we have an index file called index.ndx. Group 10 now containts the loop and group 11 the rest of the protein which should remain untouched.
Now we run tdist with the loop input file loop.cpf.
tdist -s 1ugu_min.pdb -pos index.ndx -flex index.ndx -inp loop.cpf > now choose group 11 for fixed positions and group 10 for flexible residuesThis will generate a topology with position constraints for group 11 and for group 10 only primary structure constraints will be defined. The command
tdisco -n 10 -cfreq 2000 -op -nofitwill generate 10 alternate loop conformations and write each structure to a pdb file.
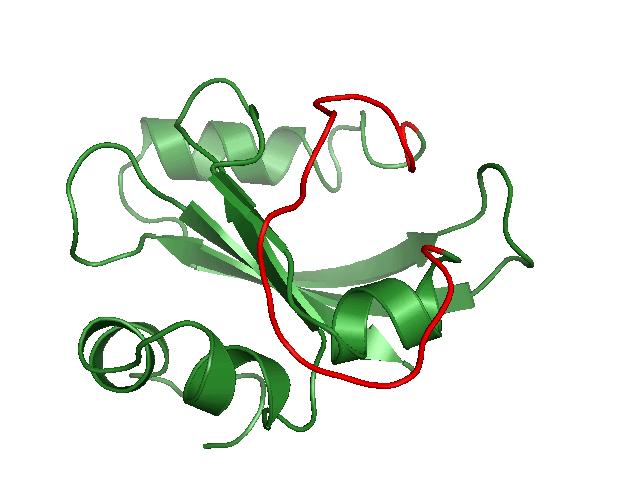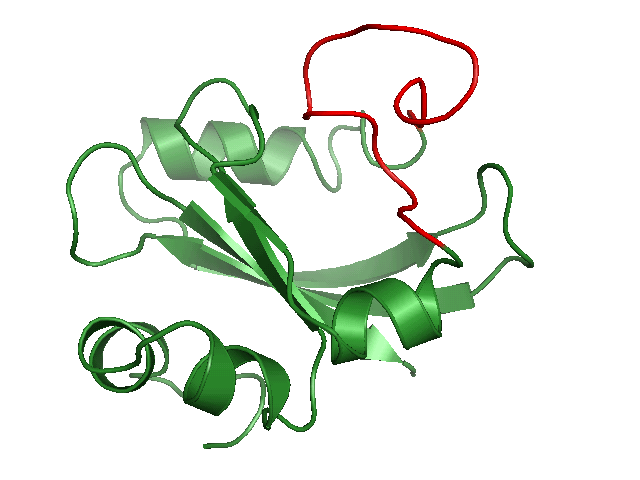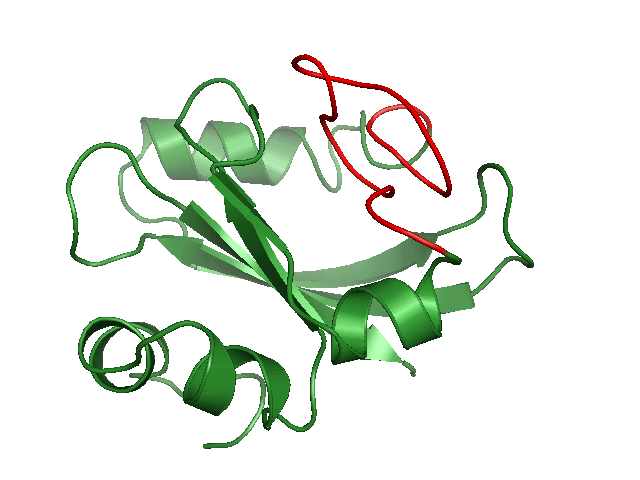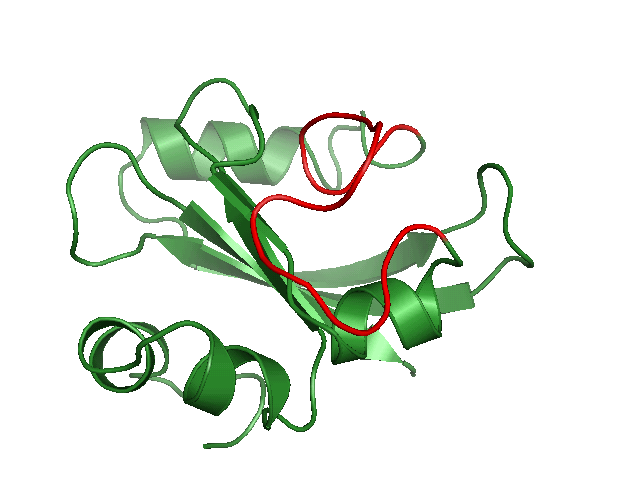
Generating conformational ensembles of ligands/compounds
tCONCOORD can also generate conformational ensembles of small organic molecules. Structural constraints (rotatable bonds/planar groups) are estimated from the input structure and default parameters are used as simulation parameters (VDW radii etc.) To generate such an ensemble, simply switch of all constraint settings in the parameter file as in compound.cpf. To examplify the procedure we generate an ensemble of a compound from the ZINC database (http://zinc.docking.org/). Download the compound ZINC00000018_u.pdb. Make sure that we have unique atom names in the pdb file ("C1"..."Cn" instead of n times "C"). Afterwards you can run tdist using$ tdist -s ZINC00000018_u.pdb -inp compound.cpfand with
$ tdisco -n 100 -on ensemble.pdb100 structures are generated and written as pdb trajectory file.