Protein stability calculations using alchemical free energy simulations (Biophys. J. (2010), accepted)
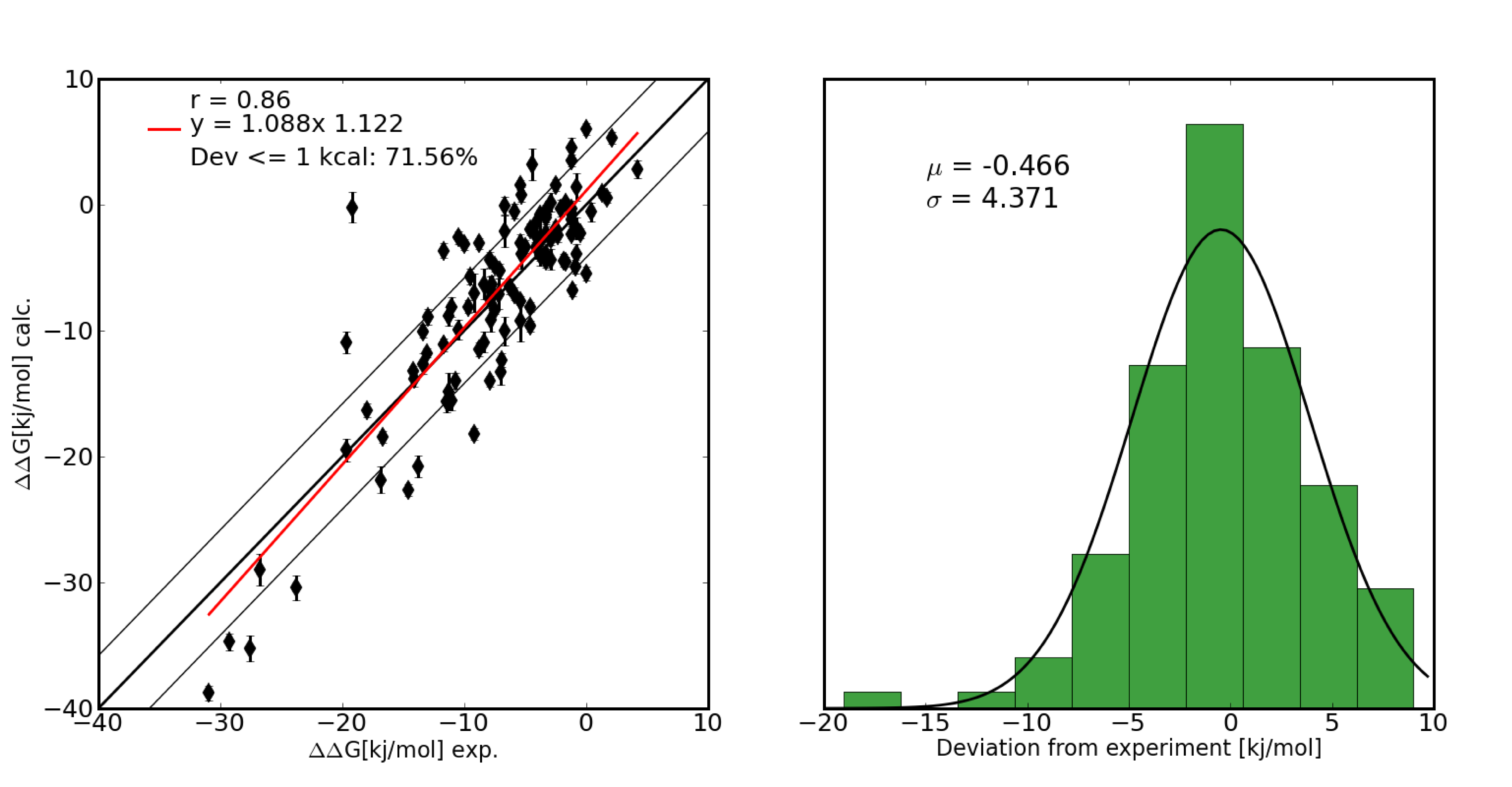
Thermodynamic stabilization of proteins is of great importance for industrial and pharmaceutical applications. The ability to accurately calculate the thermodynamic consequences of point mutations is therefore a prerequisite for rational protein engineering. We have developed a computational framework to automate the setup of lambda-based free energy calculations for all amino acid mutations occuring in natural proteins. A large set of point mutations in the well studied enzyme Barnase were calculated and quantitative agreement with experimental data was obtained.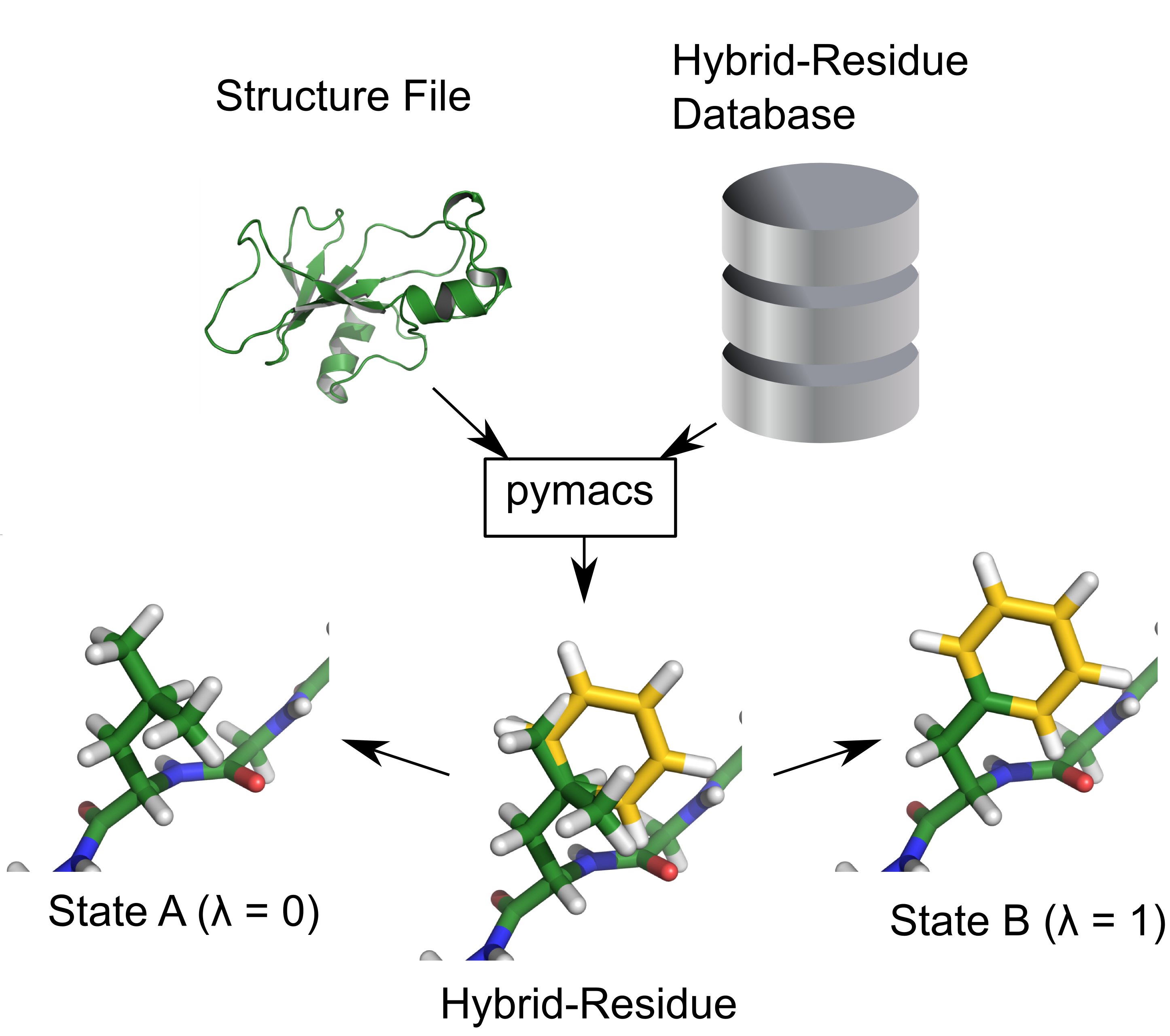
Holo structure prediction from apo-conformations (PLoS Comput Biol, 6,1:e1000634 (2010))
Many proteins undergo large conformational changes upon ligand binding. Structure determination of protein/ligand complexes however, is often difficult in such cases. Since virtual screening to apo proteins usually yields poor enrichment factors we developed a protocol to predict receptor structures in the ligand-bound state based solely on their unbound (apo) conformation, a known ligand and relatively easy to obtain experimental data (e.g. radius of gyration from SAXS). Using this protocol we were able to predict close to native ligand-bound conformations of proteins which undergo large domain motions up to 7.1A backbone RMSD. For 8 of 10 cases we were also able to predict the ligand binding mode with an accuray within 3A RMSD.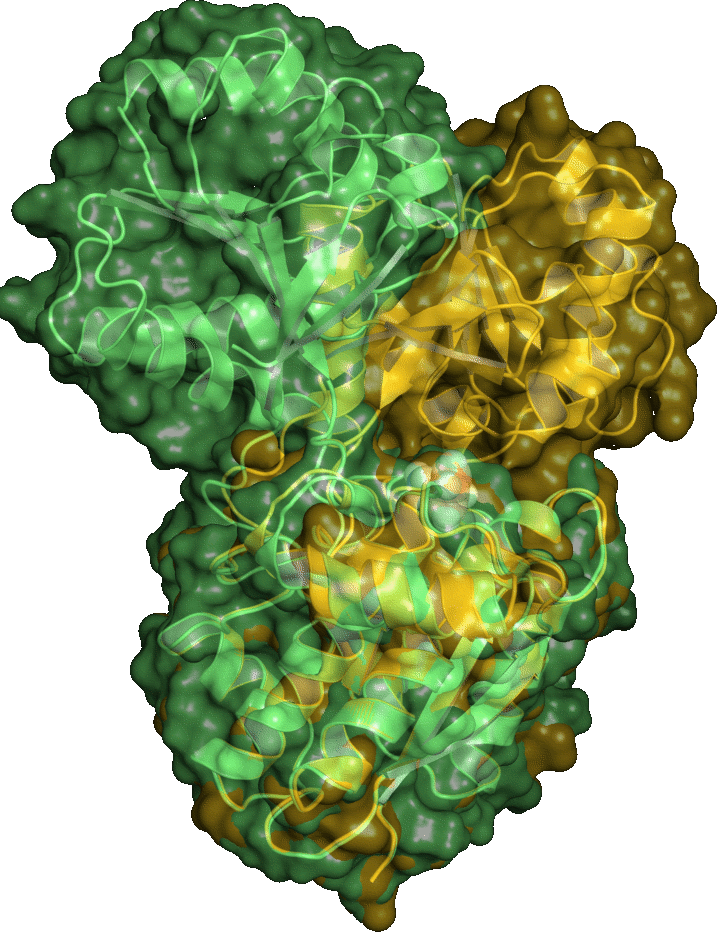
Geometry-based Sampling of Conformational Transitions in Proteins (Structure 15:1482-1492 (2007))
Proteins often undergo conformational changes to fulfil their function ranging from small side-chain movements to complete domain reorientations or partial unfolding and refolding. Large transitions routinely require crossing high energy barriers, which puts them far out of reach for beeing studied with Molecular Dynamics simulations.We use a geometry-based approach (tCONCOORD) to predict ensembles, which contain structures in different conformational states, regardless of energy barriers. The method rests on a thorough analysis of a given input structure. Hydrogen bonds, hydrophobic clusters and topological constraints (bonds, angles, planarities, chiralities) are turned into geometrical constraints, which can be regarded as a construction plan of the protein.
In a second step, structures are built from random coordinates based on the predefined constraints using the CONCOORD-algorithm. The resulting ensemble (typically 100+ structures) can be used to analyze essential degrees of freedom, to identify rigid and flexible parts in protein structures or to obtain starting points for MD-simulations from different conformations.
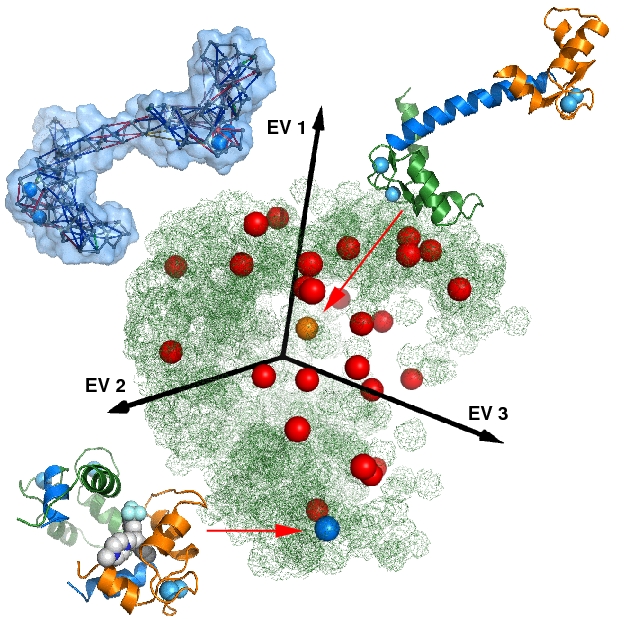
Figure. Conformational Sampling of Calmodulin: The picture shows a 3D-PCA. X-ray structures (blue and orange dots), a NMR-ensemble (red dots) and a tCONCOORD-ensemble (green cloud) are projected on the first 3 principal components. The tCONCOORD ensemble, using the acivated conformation (upper right) as input, samples the complete conformational space of the experimentally determined structures. The best match to the ligand bound state of calmodulin (lower left) is only 2.8 A.
Packing Properties of Protein Structures (Proteins. 68:565-601 (2007))
Here, we have developed an approach to quantitatively determine the packing efficiency of a large set of protein structures at different levels of resolution. A "packing score" is introduced that allows a robust assessment of the degree of packing efficiency, resting on a set of atomic radii derived from a set of high resolution protein structures. The distribution of close contacts and overlaps in protein structures is invariant and highly conserved in high-resolution X-ray structures, regardless of function, size and secondary structure.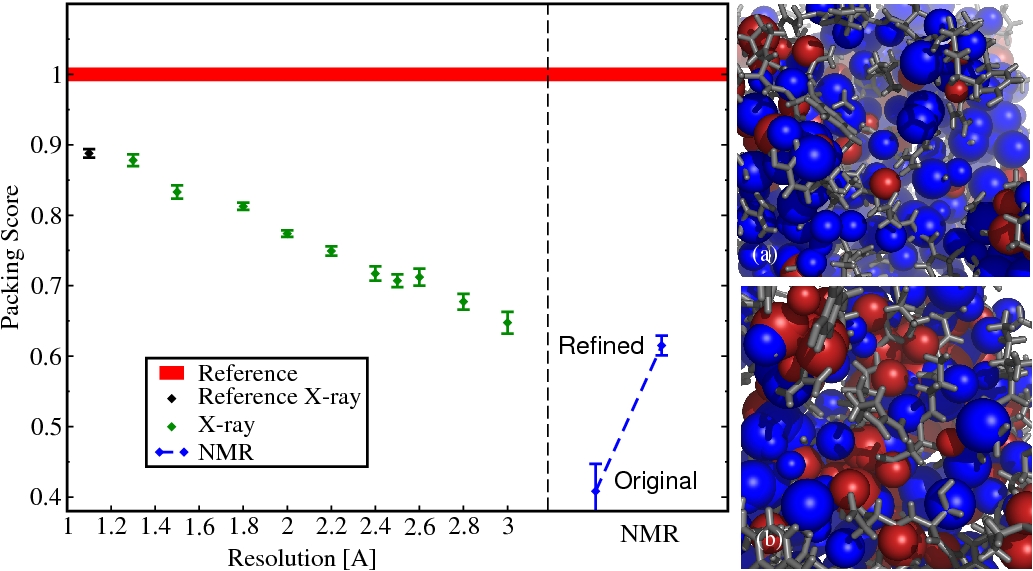
Figure. Atomic Packing in Protein Structures. Left panel: Packing scores. Red line (reference): line thickness represents the standard deviation; black: the ensemble of high resolution structures that were used to derive the atomic radii; green: X-ray structures at different levels of resolution; blue: Ensemble of NMR-structures original from the PDB and the same structures from the DRESS database. Right panel: Two structures of staphylococcal nuclease. (a) PDB 1ey4, resolved by X-ray crystallography (Resolution, 1.6 ). (b) PDB 1jor, resolved by NMR. The blue colored atoms are well packed and embedded in their local environment. Red colored atoms cause overlaps with their neighbors.