tCONCOORD:
Introduction:
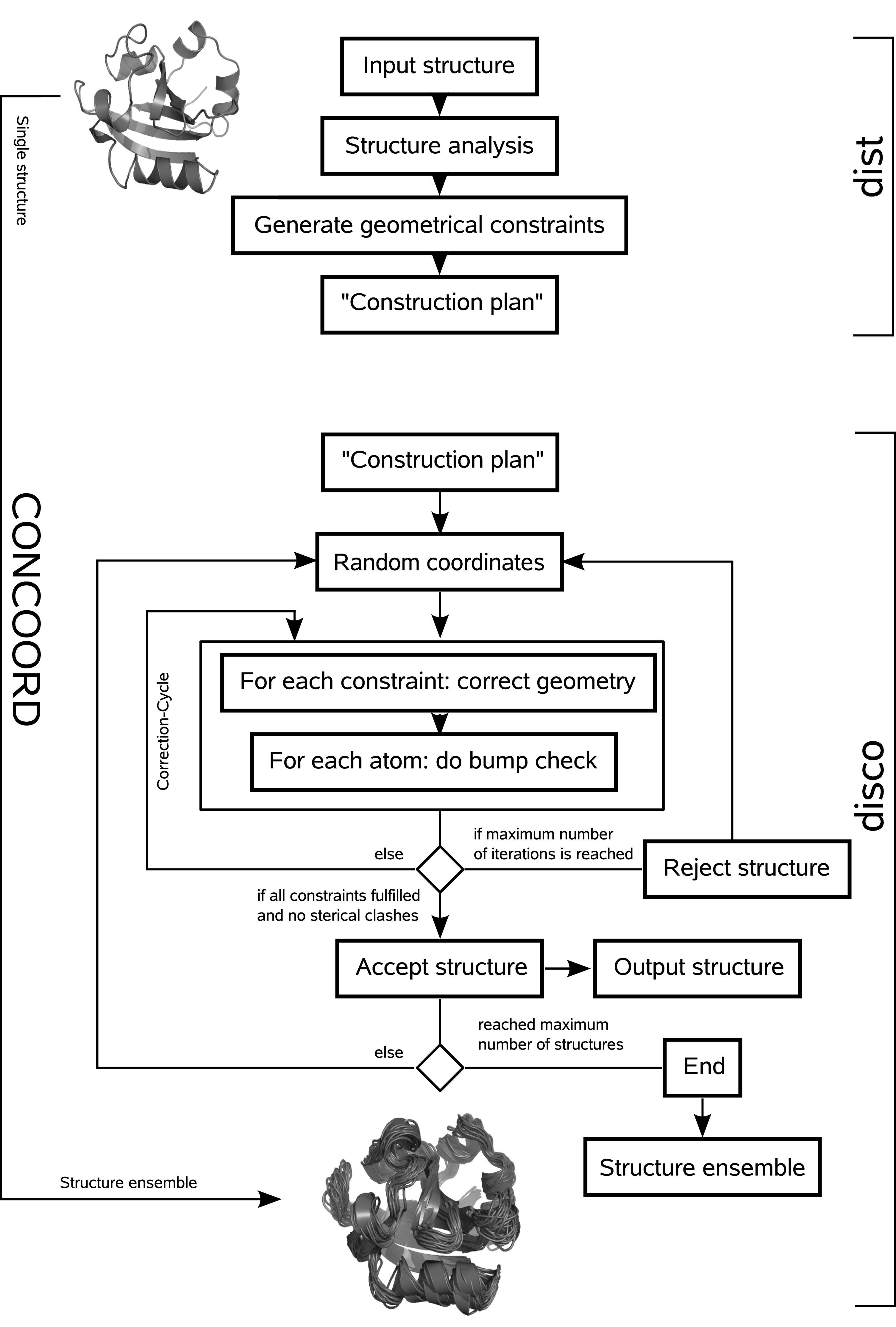
tCONCOORD predicts protein conformational flexibility based on geometrical considerations. In a first step, the protein structure is analyzed and turned into a set of constraints, mostly distance constraints but also angle, chiral and planarity constraints with upper and lower bounds. This set of constraints serves as a kind of construction plan for the protein.In a second step, the protein structure is rebuilt based on the predefined constraints. Starting from random atomic coordinates the constraints are fulfilled iteratively and a new structure is "constructed". Repeating this procedure several times leads to an ensemble of structures which represents a good sampling of the conformational space.
Download and Installation:
Download tCONCOORD-1.0_linux32bit.tgz or download tCONCOORD-1.0_linux64bit.tgz.$ tar -zxvf tCONCOORD-1.0_linuxXXbit.tgz $ source tCONCOORD-1.0/tCONCOORDRC.csh (or tCONCOORDRC.bash)tCONCOORD makes extensive use of the Gromacs package, so you must have a working gromacs installation in your system and the environmental variable GMXLIB must be set.
How to set up a tCONCOORD run:
tCNC needs a single structure file (protein.pdb) and a parameter file (input.cpf) as input. The program tdist generates geometrical constraints using the command:tdist -s protein.pdb -inp input.cpf
Structures are generated using the program tdisco.The command
tdisco -n 100 -opwill generate 100 structures and output each structure in pdb format.
Stucture preparation:
tCONCOORD has been parametrized and mainly tested for all atom simulations. Therefore, I recommand to use fully protonated structures. The structure analysis carried out by tdist will not work properly if not at least polar hydrogens are present. Furthermore I recommand to do a short energy minimization before running tCONCOORD to get rid of the worst clashes.Constraint definition with tdist:
tCONCOORD takes the information required for building structures from two sources. The first source is the input structure from which tCONCOORD derives the topology (bonds, angles, ...) and also hydrogen bonds and hydrophobic clusters if recommanded. You may influence the way how tCONCOORD defines constraints in different ways. Either you change setting in the parameter file or from the command line.The command line options are:
-s : input, structure file (pdb,gro,g96,tpr....) tCONCOORD defines bonds and angles from the input structure. However, if a tpr file is used as input, tCONCOORD takes these information from the interaction definition in the tpr file. -inp : input, parameter file -od : output, file with distance and angle constraints -op : output, pdb file -top: output, tCONCOORD topology file In this file, information about bonds, angles, chiral groups, planar groups..... are stored. -log: output, log file -clean: output, pdb file tCONCOORD does some renaming and renumbering of atoms and residues. You may use this option to clean your input file. NOTE: No constraint definition is performed when using this option. -grp: input, gromacs index file tCONCOORD performs a network analysis in order to identify "relatively rigid" regions in the structure. You can omit this procedure and define your own "relatively rigid" regions. NOTE: If you want some parts to be completely rigid use position constraints -pos: input, gromacs index file for defining position constraints Atoms with position constraints remain untouched. -flex: input, gromacs index file for defining flexible groups Use this option if you want a part of the structure to be fully flexible. tCONCOORD will only define primary structure constraints for these atoms. -tidx: input, gromcas index file for target definition ... to be done.... -ef: input, exclusions and forced constraints Use this option if you want certain interactions to be switched off or to apply artificial constraints. -add: input, define bonds -h: print help message and exit -def: define how many constraints should be defined (choose value between 0 and 1) -seed: initial random seed (-1 means make a seed) -merge: number of common group members for group fusion -H: remove all hydrogens -pH: remove all non-polar hydrogens -ign: always use the lib file entries for constraint definition Usually, upper and lower bounds are adapted to match the input structure.
The tCONCOORD parameter file (.cpf):
In the cpf file you can define change a number of options for the constraint definition. These options are:Structure generation:
Structures are generated by the program tdisco. Command line options are:-top: input, topology file (.ctp) generated with tdist -x: output, trajectory file in .xtc format -on: output, trajectory file in .pdb format -op: output, write each structure in pdb format -grp: input, gromacs index file Instead of starting from completely randomized atom coordinates you may also select a subset of the structure to be randomized. -ref: output, reference structure in pdb format -log: output, log file -target: ... to be done.... -tidx: .... to be done... -h: print help message and quit -v: be verbose -n: number of strutcures to be generated -i: maximum number of iterations -nsteps: maximum number of refinement steps -start: start numbering with # -rand: start from random coordinates -pert: only perturb starting configuration instead of complete randomization -seed: initial random seed -nu: maximum number of unsuccessful trials -check: check bounds -cfreq: check frequency tdisco regularly checks for improvement in the structure generation process. For slow converging cases you may change the check frequency. -refine: do refinement steps -fit: fit generated structures to the input reference -btol: bump tolerance -damp: damp bounds useful for slow converging cases