
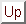

Generally, a coarse-grained effective description of a (microcanonical)
system with a Hamiltonian
 , where
, where
 denote the
3N Cartesian coordinates of the
N particles within the system and
denote the
3N Cartesian coordinates of the
N particles within the system and
 their momenta, can be achieved by explicitly considering
the dynamics of only a few degrees of freedom
their momenta, can be achieved by explicitly considering
the dynamics of only a few degrees of freedom  ,
commonly referred to as
`conformational coordinates'[65].
If the remaining degrees of freedom are regarded as a
heat bath, the resulting system of reduced dimension
belongs to a canonical ensemble.
,
commonly referred to as
`conformational coordinates'[65].
If the remaining degrees of freedom are regarded as a
heat bath, the resulting system of reduced dimension
belongs to a canonical ensemble.
The free energy landscape of that sub-system is
a potential of mean force[66],  , which
determines, together with the heat bath, the dynamics of the
conformational coordinates
, which
determines, together with the heat bath, the dynamics of the
conformational coordinates  . By means of the
canonical (projected) phase space density
. By means of the
canonical (projected) phase space density  :
:
 can be derived from
the phase space density
can be derived from
the phase space density  generated by
the dynamics of the entire system:
generated by
the dynamics of the entire system:
As an illustration, Figure  shows a contour-plot of the free energy landscape of our protein
model, which has been determined from
shows a contour-plot of the free energy landscape of our protein
model, which has been determined from
 according to
(). Here,
the two distances
according to
(). Here,
the two distances  and
and
 between
atoms #12 and #36 as well as #12 and #87, respectively,
have been chosen as conformational coordinates
(cf. Fig. ).
The canonical ensemble, from which we derived
between
atoms #12 and #36 as well as #12 and #87, respectively,
have been chosen as conformational coordinates
(cf. Fig. ).
The canonical ensemble, from which we derived  ,
has been generated from the
complete set of 232 1-ns-trajectories by recording both distances every
8 fs in the course of the simulations. Discrete values for
,
has been generated from the
complete set of 232 1-ns-trajectories by recording both distances every
8 fs in the course of the simulations. Discrete values for  have then been
determined on a grid of
have then been
determined on a grid of  resolution by computing a
two-dimensional histogram using square bins of
resolution by computing a
two-dimensional histogram using square bins of  side length.
This choice represents a compromise
between the conflicting aims of high
resolution and low statistical fluctuations of frequency counts per bin.
The contour plot in Fig. represents a smoothened
version of the two-dimensional histogram.
side length.
This choice represents a compromise
between the conflicting aims of high
resolution and low statistical fluctuations of frequency counts per bin.
The contour plot in Fig. represents a smoothened
version of the two-dimensional histogram.

Figure: Contour-plot of the free energy landscape
 derived from a projection of the phase space
density
derived from a projection of the phase space
density  onto two conformational coordinates,
namely the distances between atoms #12 and #36, and atoms #12 and #87,
respectively (cf. Fig. ).
The inset shows the values of the energy
landscape in units of
onto two conformational coordinates,
namely the distances between atoms #12 and #36, and atoms #12 and #87,
respectively (cf. Fig. ).
The inset shows the values of the energy
landscape in units of  along a hypothetical reaction
coordinate (bold line) connecting
the points A-B-C-D-B-E.
along a hypothetical reaction
coordinate (bold line) connecting
the points A-B-C-D-B-E.
The inset of Fig. illustrates the shape of the
free energy landscape W
by plotting its value in units of  (at T=300K)
along an arbitrarily chosen `reaction coordinate'
(bold line) passing through the three minima. W has been interpolated using a
cubic spline function.
(at T=300K)
along an arbitrarily chosen `reaction coordinate'
(bold line) passing through the three minima. W has been interpolated using a
cubic spline function.
The energy landscape in Fig. exhibits
three distinct minima, denoted as `B', `C', and `D',
respectively.
As suggested on the basis of experimental data by
Frauenfelder[4] such regions of low free energy
in conformational space, separated by free energy barriers,
generally define distinct conformational substates of a protein.
Accordingly, we define our model to be in substate `B', `C', or `D',
respectively, if its conformation lies in the corresponding region
of the free energy landscape.
Like the Brownian motion of a particle
coupled to a heat bath,
the dynamics of the system within
the energy landscape shown in Fig. is diffusive.
Occasionally, the fluctuating forces
generated by the heat bath drive the system across one of the
energy barriers and induce a conformational transition,
which reveals itself as a rapid change in the sterical structure
of the model. Such sudden structural transitions are, e.g.,
apparent in the lower parts of Fig. .
Note, that the above definition of conformational substates
differs from the approach commonly employed for
theoretical explorations of
substate hierarchies[67,68]. In these studies,
the distribution of
thermally accessible local minima of potential energy
within configurational space is studied, and
it is assumed, that these local minima or clusters thereof
can provide information
on the distribution of conformational substates. At low temperatures,
where entropic contributions are small and safely can be neglected,
the potential energy landscape definitely can
serve as a tool for the analysis of conformational substates.
However, at room temperature the suggested relation between
accessible minima of potential energy and conformational substates,
defined as minima of free energy, is questionable.
In contrast, our approach allows
the study of conformational substates at physiological temperatures,
as it refers to the free energy landscape within conformational space.
Therefore,
if applied to realistic protein models, our approach enables
comparisons of theory and experiment.
Admittedly, much larger computational effort is involved in such analysis,
because a sufficiently dense sampling of phase space by extended
simulations is required for the determination of  .
At present, that computational effort restricts our method to studies of
simplified protein models.
.
At present, that computational effort restricts our method to studies of
simplified protein models.
As can be seen in Fig. , the conformational substates
of our protein model are stable on the time scale of several ten
picoseconds; conformational transitions occur on scales above
a few hundred picoseconds. In order to study, whether memory effects are
present in the dynamics on these two time scales,
we will now consider two simple stochastic models.
The first model will refer to and will be derived from
the protein dynamics within one particular conformational substate.
We choose a one-dimensional Langevin model, which describes the
Brownian motion of a particle in a potential of mean force,  .
We will derive this effective potential from the simulation employing the
same procedure that was used to compute the free energy landscape shown
in Fig. .
The second model serves to describe the conformational dynamics of
transitions between the three conformational substates apparent
in Fig. on the longer time scale above several hundred
picoseconds. We will choose a Markov model which neglects the protein dynamics
within each substate.
.
We will derive this effective potential from the simulation employing the
same procedure that was used to compute the free energy landscape shown
in Fig. .
The second model serves to describe the conformational dynamics of
transitions between the three conformational substates apparent
in Fig. on the longer time scale above several hundred
picoseconds. We will choose a Markov model which neglects the protein dynamics
within each substate.

