Current Projects
Alchemical free energy calculations
Potassium channels: Mechanisms of ion conduction, selectivity and gating
Free energy calculations for protein design
Therapeutics and diagnostics for neuropathological protein aggregates
NMR based structure determination
Collective functional dynamics
Alchemical free energy calculations
In this project we are working on the method development and application of the approaches to calculate free energy differences by exploiting non-physical, alchemical, pathways across a thermodynamic cycle. In a number of applications we have been able to demonstrate a remarkable predictive accuracy of our methods in assessing changes in protein thermostability upon amino acid mutation. Further steps are made towards an accurate first principles based protein-protein and protein-peptide binding free energy estimation. Another branch of interest comprises protein-ligand interactions and automated alchemical calculation of the free energy differences for ligand modifications.
References:
[1] Vytautas Gapsys, Laura Perez-Benito, Matteo Aldeghi, Daniel Seeliger, Herman van Vlijmen, Gary Tresadern, Bert L de Groot. Large scale relative protein ligand binding affinities using non-equilibrium alchemy. Chemical Science. 11:1140-1152 (2020)
[2] Matteo Aldeghi, Bert L de Groot, Vytautas Gapsys. Accurate Calculation of Free Energy Changes upon Amino Acid Mutation. in Computational Methods in Protein Evolution. 19-47 (2019)
[3] Vytautas Gapsys, Bert L de Groot. pmx Webserver: A User Friendly Interface for Alchemistry. J. Chem. Inf. Model. 57: 109-114 (2017).
[4] Vytautas Gapsys, Servaas Michielssens, Daniel Seeliger, and Bert L. de Groot. pmx: Automated protein structure and topology generation for alchemical perturbations. J. Comput. Chem. 36:348-354 (2015)
[5] Vytautas Gapsys, Servaas Michielssens, Jan Henning Peters, Bert L. de Groot, Hadas Leonov. Calculation of Binding Free Energies. Molecular Modeling of proteins: 2nd edition. Book Series: Methods in Molecular Biology 1215: 173-209 (2015)
[6] Vytautas Gapsys, Daniel Seeliger, and Bert L. de Groot. New Soft-Core Potential Function for Molecular Dynamics Based Alchemical Free Energy Calculations. J. Chem. Theor. Comput. 8:2373-2382 (2012)
Vytautas Gapsys, Matteo Aldeghi
Potassium channels: mechanisms of ion conduction, selectivity and gating
All animal cells are characterized by steep electrochemical gradients of Na+ and K+ ions across the plasma membrane. Potassium channels facilitate the passage of K+ ions to near-diffusion limited rates, while reliably excluding smaller Na+ ions. A clear separation between K+ and Na+ currents is necessary to ensure sharp action potentials, facilitating the rapid propagation of electric signals in excitable cells such as neurons. In this project, we are aiming at elucidating the molecular mechanism of ion conduction of different ion through representative potassium channels (e.g. KcsA, MthK, Kv1.2) and their effect on spontaneous gating transitions. We are using the computational electrophysiology approach that enables us to directly observe hundreds of individual permeation events under voltage, closely mimicking electrophysiological recordings. A special attention is dedicated to force field calibration and validation.References:
[1] Wojciech Kopec, David A. Köpfer, Owen N. Vickery, Anna S. Bondarenko, Thomas L.C. Jansen, Bert L. de Groot, Ulrich Zachariae Direct knock-on of desolvated ions governs strict ion selectivity in K+ channels. Nature Chemistry 10:813-820 (2018).
Lipid-Protein Interaction in Potassium Channel Gating and Permeation. The potassium channels are widely distributed highly selective ion channels gated by diverse physiological signals. Due to the vast lipid species constituting cell membranes, and their spatial and temporal heterogeneous distribution, membrane bilayers are known to impose strong effects on protein structure and function. A variety of experiments have suggested effects of lipid-protein interaction on channel conductance. For instance, negatively charged lipids not only regulate channel gating, but also accelerate ion permeation by polarizing local environment at the membrane-solvent interface. We use molecular dynamics simulations, in particular the computational electrophysiology simulation method, to investigate influence of general membrane properties on ion permeation of open state channels. At present, we are testing two membrane properties: the bilayer thickness and membrane surface charge. These simulations closely mimick electrophysiology experiments. This project allows insight into the response of protein function to membrane general properties and the underlying mechanism at atomistic level, in the case of potassium channels.
Free energy calculations for protein design
Applications of free energy calculations in biophysics, drug and protein design Proteins are involved in all fundamental processes of living organisms and have evolved into extremely precise and efficient machines. Thanks to their versatility, protein-based materials have the potential to tackle many challenges in biomedicine and nanotechnology. In this project, we aim to assess the feasibility of using alchemical free energy calculations for the optimisation of proteins with specific target functions [1]. As protein mutations also play an important role in the development of drug resistance, we are also exploring the potential of free energy calculations for the prediction of resistance. Finally, we use the thermodynamic cycles that one can build with alchemical free energy calculations as a flexible tool to study how mutations affect processes such as protein folding, ligand-protein and protein-protein binding in various systems of interest [2].References:
[1] M. Aldeghi, V. Gapsys, B.L. de Groot "Accurate estimation of ligand binding affinity changes upon protein mutation. ACS Cent. Sci. 2018, 4(12), 1708-1718
[2] A.W. Yee*, M. Aldeghi*, M. Blakeley, A. Ostermann, P. Mas, M. Moulin, D. de Sanctis, M.W. Bowler, C. Mueller-Dieckmann, E. Mitchell, M. Haertlein, B.L. de Groot, E. Boeri Erba, V.T. Forsyth. A molecular mechanism for transthyretin amyloidogenesis. Nat. Commun. 2019, 10, 925
Therapeutics and diagnostics for neuropathological protein aggregates
The di-phenyl-pyrazole compound anle138b is a known inhibitor of oligomeric aggregate formation in vitro and in vivo. Therefore, anle138b is considered a drug candidate for a number of neurodegenerative diseases in humans. At the same time, anle138b provides a promising scaffold for positron emission tomography tracer design. Here, we set out to elucidate anle138b's binding to oligomeric aggregates of amyloidogenic proteins, as well as fibrils using all-atom molecular dynamics simulations. We also explore the atomistic details of aggregation mechanisms and the potential neurotoxic mode of action of small nonfibrillar aggregates.References:
[1] D. Matthes, V. Gaspys, C. Griesinger, B. L. de Groot. Resolving the atomistic modes of anle138b inhibitory action on peptide oligomer formation. ACS Chem. Neurosci. (2017).
[2] Antonschmidt, L., Matthes, D., Dervisoglu, R. et al. The clinical drug candidate anle138b binds in a cavity of lipidic α-synuclein fibrils. Nat. Commun. (2022).
[3] Matthes, D., B. L. de Groot. Molecular dynamics simulations reveal the importance of amyloid-beta oligomer β-sheet edge conformations in membrane permeabilization. J. Biol. Chem. (2023).
Collective functional dynamics
Nonadditivity of correlated amino acid mutations. Interactions between amino acid mutations play a key role in protein engineering regarding the understanding of functional mechanisms such as cooperativity and allostery. The effect of a double mutation on properties like thermostability or binding affinity can be significantly different from the sum of effects of the separate single mutations. In this case the mutations are correlated which is revealed by a nonadditivity of the corresponding free energies. While such a coupling is expected for residues in close spatial contact due to the formation of directed interactions as hydrogen bonds or salt bridges a set of enigmatic cases with significant nonadditivities persisting over large distances has been identified. In this project the nonadditivity of correlated mutations are investigated via alchemical free energy calculations and protein dynamic analysis tools to unravel the underlying coupling mechanism.
Previous Projects
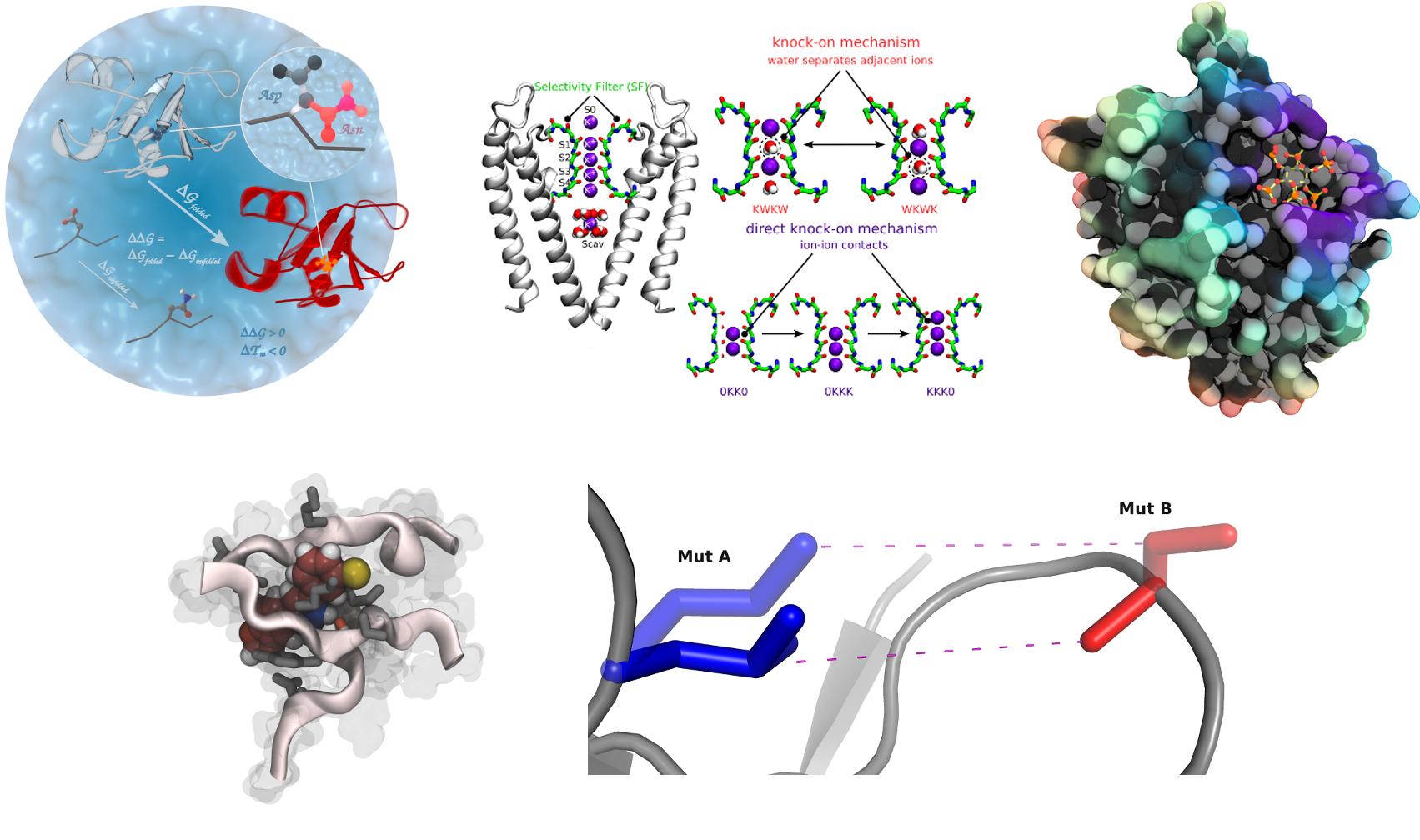
Lipid-protein Interactions
 In this project we focus on the effect of embedded peptides or proteins on
the surrounding membrane and on the effect of the membrane on embedded
peptides. Small peptides involve HIV-1 gp41 or gramicidin like channels.
Membrane channel proteins involve VDAC (Voltage Dependent Anion
Channel) [1] and Aquaporins [2] among others [3]. These systems are being
studied in different types of model membranes (DMPC, DPPC, POPE, etc).
This work is done with the close collaboration of NMR, EPR and electron
crystallography groups. Tools for analyzing L-P complexes are also been
developed [4].
In this project we focus on the effect of embedded peptides or proteins on
the surrounding membrane and on the effect of the membrane on embedded
peptides. Small peptides involve HIV-1 gp41 or gramicidin like channels.
Membrane channel proteins involve VDAC (Voltage Dependent Anion
Channel) [1] and Aquaporins [2] among others [3]. These systems are being
studied in different types of model membranes (DMPC, DPPC, POPE, etc).
This work is done with the close collaboration of NMR, EPR and electron
crystallography groups. Tools for analyzing L-P complexes are also been
developed [4].
References:
[1] Saskia Villinger, Rodolfo Briones, Karin Giller, Ulrich Zachariae, Adam Lange, Bert L. de Groot, Christian Griesinger, Stefan Becker, Markus Zweckstetter. Functional dynamics in the voltage dependent anion channel. Proc. Nat. Acad. Sci. 107: 22546-22551 (2010).
[2] C. Aponte-Santamaria, R. Briones, A.Schenk, T. Walz, B. de Groot. Molecular driving forces defining lipid positions around aquaporin-0. PNAS 109: 44319-44325 (2012).
[3] Rasmus Linser, Nicola Salvi, Rodolfo Briones, Petra Rovo, Bert L. de Groot, Gerhard Wagner. The membrane anchor of the transcriptional activator SREBP is characterized by intrinsic conformational flexibility. Proc. Nat. Acad. Sci. 112: 12390-12395 (2015).
[4] Vytautas Gapsys, Bert L. de Groot, Rodolfo Briones. Computational analysis of local membrane properties. J. Comput. Aided Mol. Des. 27: 845-858 (2013).
[5] Rodolfo Briones, Camilo Aponte-Santamaria, Bert L de Groot. Localization and ordering of lipids around Aquaporin-0: Protein and lipid mobility effects Front. Physiol. 8: 124 (2017).
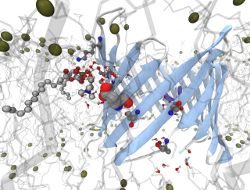
Insight into Ion Channel Gating by Computational Electrophysiology Simulations
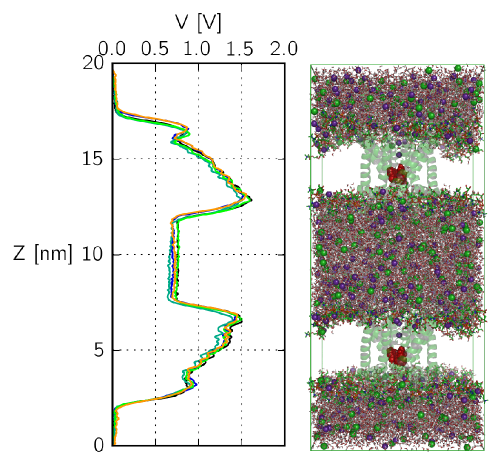 Insight into the functional mechanisms of ion channels is of
universal importance to enable drug design on the class of
membrane proteins, and to enhance our understanding of some of the fundamental
features of cells.
We have recently developed a simulation protocol called Computational
Electrophysiology
(CompEL) [1], which facilitates the atomistic simulation of ion channels in
action.
The computational electrophysiology allows to simulate membrane embeded
proteins in two compartments separated by lipid bilayers
with a sustained ion flux.
Thus far we have used CompEL to study several different membrane proteins
ranging from highly ion-selective, narrow
channels to wide diffusion pores [2-6].
Insight into the functional mechanisms of ion channels is of
universal importance to enable drug design on the class of
membrane proteins, and to enhance our understanding of some of the fundamental
features of cells.
We have recently developed a simulation protocol called Computational
Electrophysiology
(CompEL) [1], which facilitates the atomistic simulation of ion channels in
action.
The computational electrophysiology allows to simulate membrane embeded
proteins in two compartments separated by lipid bilayers
with a sustained ion flux.
Thus far we have used CompEL to study several different membrane proteins
ranging from highly ion-selective, narrow
channels to wide diffusion pores [2-6].
References:
[1] Carsten Kutzner, Helmut Grubmüller, Bert L. de Groot, Ulrich Zachariae. Computational Electrophysiology: The Molecular Dynamics of Ion Channel Permeation and Selectivity in Atomistic Detail. Biophys. J. 101: 809-817 (2011).
[2] Jan-Philipp Machtens, Rodolfo Briones, Claudia Alleva, Bert L. de Groot, Christoph Fahlke. Gating Charge Calculations by Computational Electrophysiology Simulations. Biophys. J. 112: 1396-1405 (2017).
[3] Carsten Kutzner, David A. Köpfer, Jan-Philipp Machtens, Bert L. de Groot, Chen Song, Ulrich Zachariae. Insights into the function of ion channels by computational electrophysiology simulations. BBA Biomembranes 1858:1741-1752 (2016).
[4] Jan-Philipp Machtens, Daniel Kortzak, Christine Lansche, Ariane Leinenweber, Petra Kilian, Birgit Begemann, Ulrich Zachariae, David Ewers, Bert L. de Groot, Rodolfo Briones and Christoph Fahlke. Mechanisms of Anion Conduction by Coupled Glutamate Transporters. Cell 160:542-553 (2015).
[5] David A. Köpfer, Chen Song, Tim Gruene, George M. Sheldrick, Ulrich Zachariae and Bert L. de Groot. Ion permeation in K+ channels occurs by direct Coulomb knock-on. Science 346: 352-355 (2014).
[6] Chen Song, Conrad Weichbrodt, Evgeniy S. Salnikov, Marek Dynowski, Björn O. Forsberg, Burkhard Bechinger, Claudia Steinem, Bert L. de Groot, Ulrich Zachariae, and Kornelius Zeth. Crystal structure and functional mechanism of a human antimicrobial membrane channel Proc. Nat. Acad. Sci. 110: 4586-4591 (2013)
David A. Köpfer, Chen Song, Ulrich Zachariae, Rodolfo Briones
Steric zipper peptide aggregation
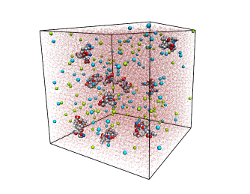
We employ molecular dynamics simulations in an explicit solvent environment to
study the spontaneous aggregation of steric zipper peptides at
atomistic detail.
These short model peptides were recently shown to
yield detailed structural insights into aggregates, known as amyloid fibrils,
which
are related to life threatening conditions in vivo (e.g. Alzheimers disease).
Our aim is to unveil the energetic and structural determinants that drive the
formation of
amyloidogenic peptide assemblies, and also stabilize the formed aggregates.
References:
[1] Dirk Matthes, Vytautas Gapsys, Julian Tim Brennecke and Bert L. de Groot. An Atomistic View of Amyloidogenic Self-assembly: Structure and Dynamics of Heterogeneous Conformational States in the Pre-nucleation Phase. Sci. Rep. 6: 33156 (2016).
[2] D. Matthes, V. Daebel, K. Meyenberg, D. Riedel, G. Heim, U. Diederichsen, A. Lange, B. L. de Groot. Spontaneous Aggregation of the Insulin-Derived Steric Zipper Peptide VEALYL Results in Different Aggregation Forms with Common Features. J. Mol. Biol., 426: 362-376 (2014).
[3] D. Matthes, V. Gapsys, B. L. de Groot, Driving forces and structural determinants of steric zipper peptide oligomer formation elucidated by atomistic simulations, J. Mol. Bio., 421: 390-416 (2012).
[4] D. Matthes, V. Gapsys, V. Daebel, B. L. de Groot, Mapping the conformational dynamics and pathways of spontaneous steric zipper peptide oligomerization, PLoS ONE, 6(5): e19129 (2011).
Dirk Matthes , Vytautas Gapsys
Thermodynamic Integration for Enhanced Protein Crystallization
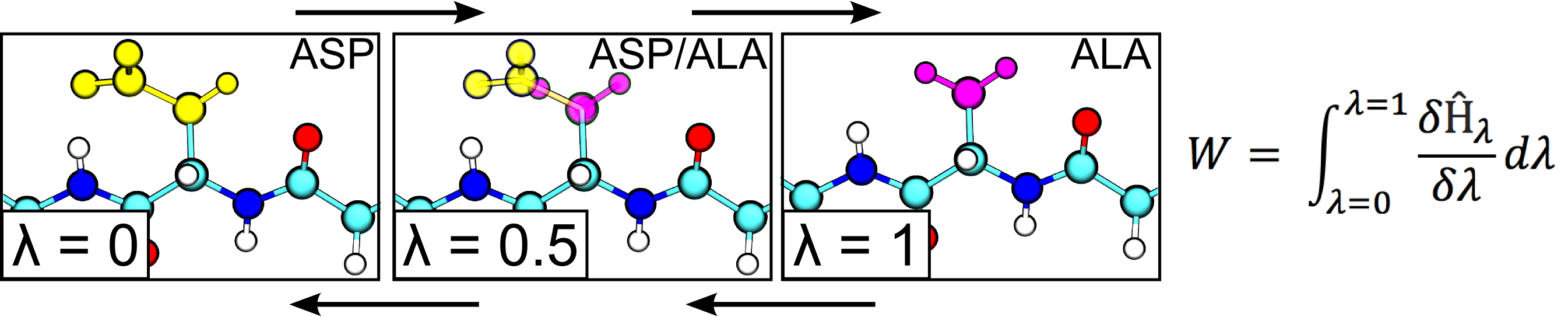
In this project aim to rigorously calculate the changes in free energy and entropy that result from protein mutations by using alchemical free energy simulations. This method allows us to enhance protein crystallization in one of two ways. First, we can selectively stabilize a specific protein conformation through mutation. Having one dominant protein structure encourages the formation of regular crystals. Second, we can mutate high-entropy surface residues to lower entropy residues. This reduces the loss in entropy during protein crystallization, thus making the process more thermodynamically favorable.
Regulation of Permeation in Aquaporins
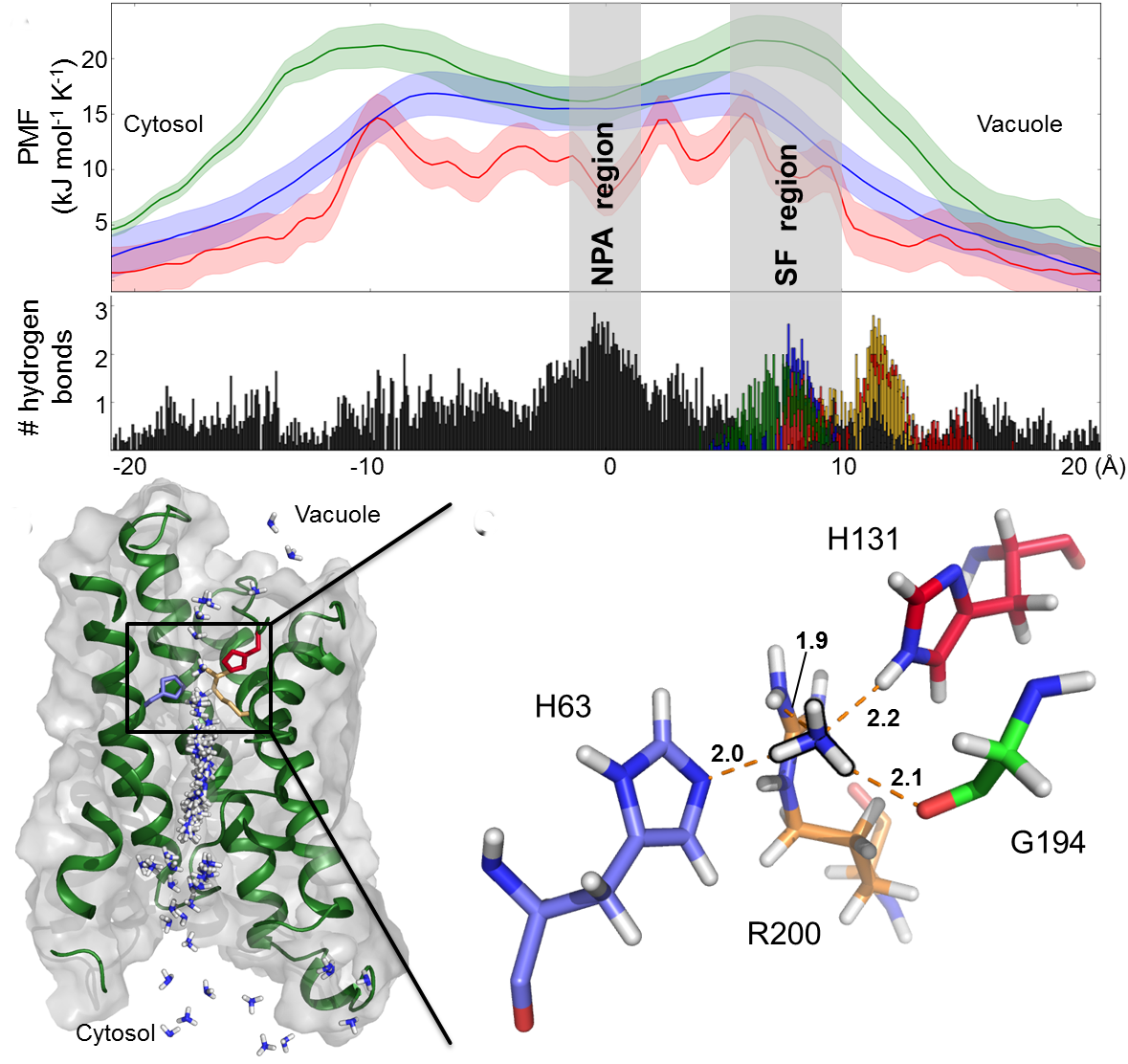 Aquaporins and related aquaglyceroporins are versatile water channels that serve
diverse purposes including the permeation of other solutes like glycerol and
ammonia. The aim of this project is to investigate the mechanisms by which aquaporins regulate the permeation
of water and alternate substrates.
In this project we use diverse techniques such as umbrella sampling, Principle Component Analysis (PCA), Functional Mode Analysis (FMA)
and Essential Dynamics to understnad how to relate the dynamics of the aquaporins structure to their function.
Aquaporins and related aquaglyceroporins are versatile water channels that serve
diverse purposes including the permeation of other solutes like glycerol and
ammonia. The aim of this project is to investigate the mechanisms by which aquaporins regulate the permeation
of water and alternate substrates.
In this project we use diverse techniques such as umbrella sampling, Principle Component Analysis (PCA), Functional Mode Analysis (FMA)
and Essential Dynamics to understnad how to relate the dynamics of the aquaporins structure to their function.
References:
[1] Shreyas Kaptan, Mette Assentoft, Hans Peter Schneider, Robert A. Fenton, Joachim W. Deitmer, Nanna MacAulay, and Bert L. de Groot. H95 is a pH-dependent gate in aquaporin 4. Structure. 23:2309-2318 (2015).
[2] Mette Assentoft, Shreyas Kaptan, Robert A. Fenton, Susan Z. Hua, Bert L. Groot, Nanna MacAulay. Phosphorylation of rat aquaporin-4 at Ser111 is not required for channel gating Glia 61: 1101-1112 (2013).
[3] Mette Assentoft, Shreyas Kaptan, Hans Peter Schneider, Joachim W. Deitmer, Bert L. de Groot, and Nanna MacAulay. Aquaporin 4 as a NH3 Channel. J. Biol. Chem. 291: 19184-19195 (2016).
[4] Andreas Kirscht, Shreyas S. Kaptan, Gerd Patrick Bienert, Francois Chaumont, Poul Nissen, Bert L. de Groot, Per Kjellbom, Pontus Gourdon and Urban Johanson. Crystal Structure of an Ammonia Permeable Aquaporin. PLOS Biol. 14:e1002411 (2016).
Shreyas KaptanUbiquitin Dynamics, Multispecificity, and Allostery
 Ubiquitin is involved in many cellular processes, most notably
proteasome-mediated proteolysis. To facilitate all of these functions, it
needs to interact with hundreds of other proteins. In order to bind these many
different molecular partners, ubiquitin must adopt different conformations. We
have previously shown that ubiquitin takes these conformations not only when
in contact with its binding partners, but also when free in solution (Lange et
al., Science, 2008), suggesting a conformational selection binding
mechanism. This conformational equilibrium can be rationally shifted to
selectively destabilize binding to particular partners (Michielssens et al.,
Angew. Chem. Int. Ed., 2014). While most of the structural rearrangements can
happen in solution prior to binding, some parts of the structure, particularly
side chains, only change upon binding (Peters and de Groot, PLOS
Comput. Biol., 2012), a process we refer to as “residual induced
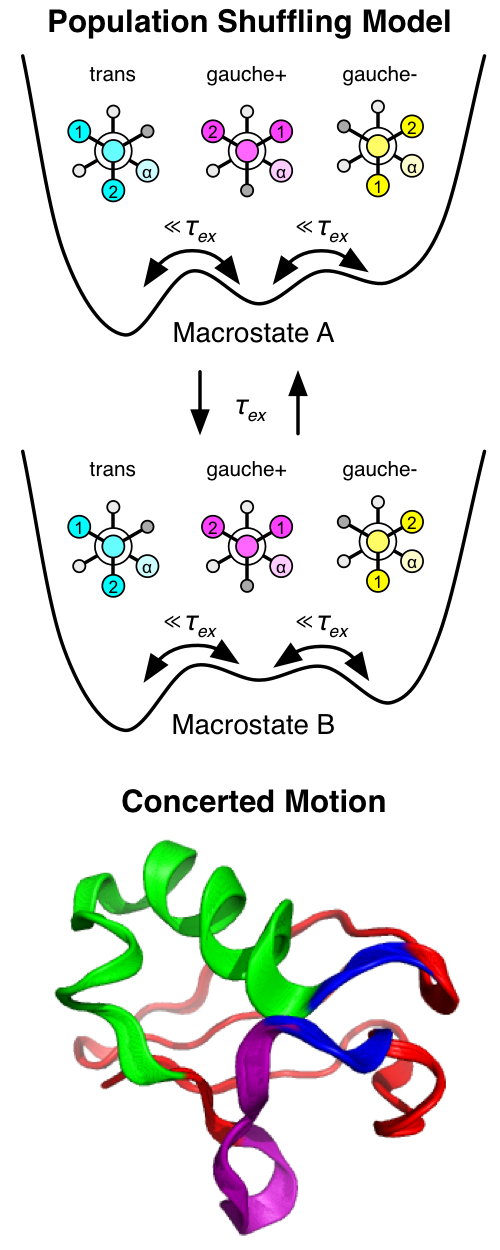
fit”. Importantly, ubiquitin side chain motions are not independent of
the backbone, but are kinetically interrelated and form a hierarchy of
timescales called “population shuffling” [1]. Through a detailed
investigation into these kinetics, we have uncovered an allosteric network
that enables multispecific binding through a conformational switch in a region
of the protein that does not directly interact with the binding partners. We
are continuing our investigations into the conformational dynamics and
kinetics of ubiquitin [2]. In particular we are interested in how subdividing structural ensembles into a hierarchy of timescales can improve agreement with experimental data and give a more accurate representation of its structural properties.
Ubiquitin is involved in many cellular processes, most notably
proteasome-mediated proteolysis. To facilitate all of these functions, it
needs to interact with hundreds of other proteins. In order to bind these many
different molecular partners, ubiquitin must adopt different conformations. We
have previously shown that ubiquitin takes these conformations not only when
in contact with its binding partners, but also when free in solution (Lange et
al., Science, 2008), suggesting a conformational selection binding
mechanism. This conformational equilibrium can be rationally shifted to
selectively destabilize binding to particular partners (Michielssens et al.,
Angew. Chem. Int. Ed., 2014). While most of the structural rearrangements can
happen in solution prior to binding, some parts of the structure, particularly
side chains, only change upon binding (Peters and de Groot, PLOS
Comput. Biol., 2012), a process we refer to as “residual induced
fit”. Importantly, ubiquitin side chain motions are not independent of
the backbone, but are kinetically interrelated and form a hierarchy of
timescales called “population shuffling” [1]. Through a detailed
investigation into these kinetics, we have uncovered an allosteric network
that enables multispecific binding through a conformational switch in a region
of the protein that does not directly interact with the binding partners. We
are continuing our investigations into the conformational dynamics and
kinetics of ubiquitin [2]. In particular we are interested in how subdividing structural ensembles into a hierarchy of timescales can improve agreement with experimental data and give a more accurate representation of its structural properties.
References:
[1] Colin A. Smith, David Ban, Supriya Pratihar, Karin Giller, Claudia Schwiegk, Bert L. de Groot, Stefan Becker, Christian Griesinger, Donghan Lee Population Shuffling of Protein Conformations Angew. Chem. Int. Ed. 54:207-210 (2015).
[2] Colin A. Smith, David Ban, Supriya Pratihar, Karin Giller, Maria Paulat, Stefan Becker, Christian Griesinger, Donghan Lee and Bert L. de Groot. Allosteric switch regulates protein-protein binding through collective motion. Proc. Nat. Acad. Sci. 113:3269-3274 (2016).
Colin Smith
Understanding the Molecular Machinery of Aquaporins through Molecular Dynamics Simulations
 Aquaporins are protein channels responsible for the permeation of water and
other solutes through biological membranes in response to osmotic
pressure. The main goal is to expand our understanding on the molecular
machinery of aquaporins by employing molecular dynamics simulations and
related computational techniques.
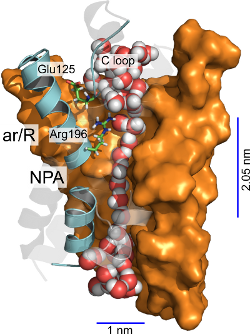
We provide a mechanism for the permeation of solutes through the Plasmodium
falciparum aquaglyceroporin, a promising antimalarial drug target. In this
mechanism, hydrophobic regions in the middle of the channel are the main water
rate limiting barriers. Furthermore, the replacement of water-arginine
interactions and solute-matching at the narrowest region of the channel are
the main determinants underlying selectivity for the permeation of solutes
like glycerol and urea (1).
We also investigate the molecular determinants governing aquaporin gating,
which has emerged as an efficient regulatory mechanism for organisms to
quickly counteract sudden osmotic shocks. Our simulations, together with
structural and functional studies, suggest that the yeast aquaporin-1 may be
gated by both serine phosphorylation or mechanosensing (2). Furthermore, we
observed voltage regulation of the single-channel water permeability of human
AQP1 and AQP4 in silico, attributed to gating transitions of the arginine
residue at the aromatic/arginine region. Our results suggests that voltage
sensitivity may be a general feature of aquaporins, a hypothesis to be tested
experimentally (3).
Aquaporins are protein channels responsible for the permeation of water and
other solutes through biological membranes in response to osmotic
pressure. The main goal is to expand our understanding on the molecular
machinery of aquaporins by employing molecular dynamics simulations and
related computational techniques.
We provide a mechanism for the permeation of solutes through the Plasmodium
falciparum aquaglyceroporin, a promising antimalarial drug target. In this
mechanism, hydrophobic regions in the middle of the channel are the main water
rate limiting barriers. Furthermore, the replacement of water-arginine
interactions and solute-matching at the narrowest region of the channel are
the main determinants underlying selectivity for the permeation of solutes
like glycerol and urea (1).
We also investigate the molecular determinants governing aquaporin gating,
which has emerged as an efficient regulatory mechanism for organisms to
quickly counteract sudden osmotic shocks. Our simulations, together with
structural and functional studies, suggest that the yeast aquaporin-1 may be
gated by both serine phosphorylation or mechanosensing (2). Furthermore, we
observed voltage regulation of the single-channel water permeability of human
AQP1 and AQP4 in silico, attributed to gating transitions of the arginine
residue at the aromatic/arginine region. Our results suggests that voltage
sensitivity may be a general feature of aquaporins, a hypothesis to be tested
experimentally (3).
References:
[1] Camilo Aponte-Santamaria, Jochen S. Hub and Bert L. de Groot. Dynamics and energetics of solute permeation through the Plasmodium falciparum aquaglyceroporin. PCCP. 12:10246-10254 (2010).
[2] Gerhard Fischer, Urszula Kosinska-Eriksson, Camilo Aponte-Santamaria, Madelene Palmgren, Cecilia Geijer, Kristina Hedfalk, Stefan Hohmann, Bert L. de Groot, Richard Neutze, Karin Lindkvist-Petersson. Crystal Structure of a Yeast Aquaporin at 1.15 Angstrom Reveals a Novel Gating Mechanism. PLoS Biology. 7: e1000130 (2009).
[3] Jochen S. Hub, Camilo Aponte-Santamaria, Helmut Grubmüller and Bert L. de Groot. Voltage-regulated water flux through aquaporin channels in silico. Biophys. J. 99:L97-L99 (2010)
[4] Camilo Aponte-Santamaria, Gerhard Fischer, Petra Bath, Richard Neutze, Bert L. Groot. Temperature dependence of protein-water interactions in a gated yeast aquaporin. Sci. Rep. 7: 4016 (2017).
Ubiquitin Dynamics in Complexes
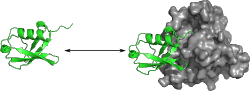 Protein-protein interactions play an important role in all metabolic
processes. However, the principles underlying these interactions are only
beginning to be understood. Ubiquitin is a small signalling protein that is
covalently attached to proteins to mark them for degradation, regulate
transport or induce other functions. As such, it interacts with and is
recognized by a multitude of binding partners.
Protein-protein interactions play an important role in all metabolic
processes. However, the principles underlying these interactions are only
beginning to be understood. Ubiquitin is a small signalling protein that is
covalently attached to proteins to mark them for degradation, regulate
transport or induce other functions. As such, it interacts with and is
recognized by a multitude of binding partners.
We use molecular dynamics simulations to investigate the effect of binding on ubiquitin by comparing simulation ensembles of ubiquitin bound to different binding partners with ensembles of unbound ubiquitin. Both collective structural behaviour and local conformational differences are being considered to identify the principles of ubiquitin binding and determine the influence of complex formation on the dynamic properties of this protein. Particularly the question of induced fit versus conformational selection scenarios both on a global and local level is investigated.
References:
[1] Servaas Michielssens, Jan Henning Peters, David Ban, Supriya Pratihar, Daniel Seeliger, Monika Sharma, Karin Giller, Thomas Michael Sabo, Stefan Becker, Donghan Lee, Christian Griesinger and Bert L. de Groot. A Designed Conformational Shift To Control Protein Binding Specificity Angew. Chem. Int. Ed. 53: 10367-10371 (2014).
[2] Jan-Henning Peters and Bert L. de Groot. Ubiquitin dynamics in complexes reveal molecular recognition mechanisms beyond induced fit and conformational selection. PLOS Comp. Biol. 8: e1002704 (2012).
Jan Henning Peters, Servaas Michielssens
Allosteric Transitions in Hemoglobin
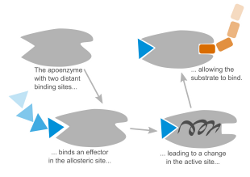
Allostery is essential for regulation in many biological systems. In allosteric systems the binding affinity of one binding site depends on the binding in a distant binding site. The information flow between these sites is assumed to be communicated through a conformational change of the system. It is still a challenging question to find this collective motion on an atomistic level. We currently focus on Hemoglobin, ABCE1 and GroEL/ES extracting collective motions from Molecular Dynamics simulations using Principal Component Analysis and related methods.
References:
[1] Martin Vesper and Bert L. de Groot. Collective Dynamics Underlying Allosteric Transitions in Hemoglobin. PLoS Comp. Biol. 9: e1003232 (2013).
Inhibition of Membrane Channel Proteins
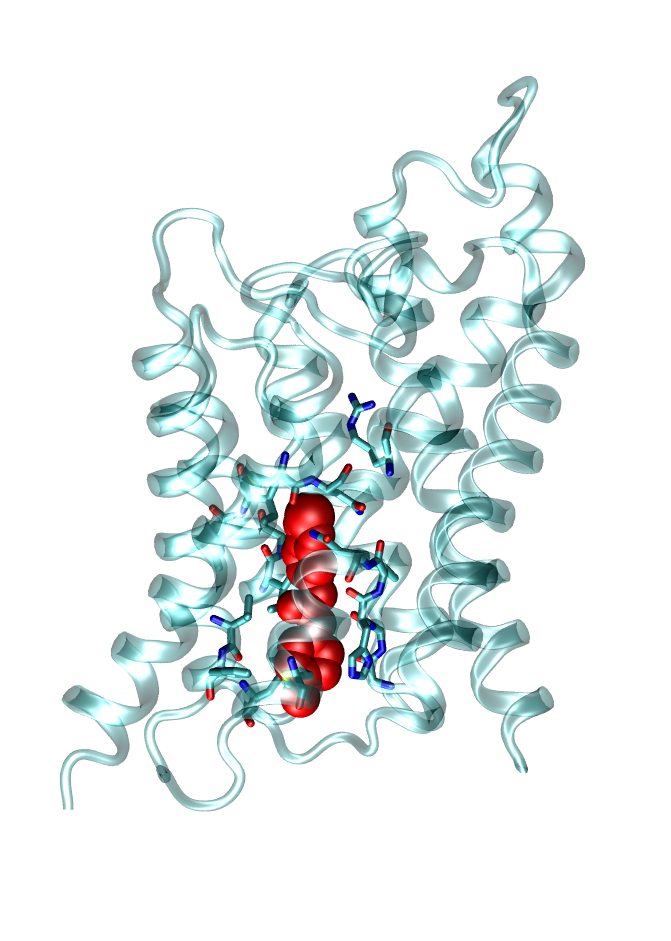 Membrane channel proteins, like ion and water channels, facilitate the permeation of water and solutes (e.g. ions) across cell membranes. They are important for many physiological processes as for example nerve conduction or the osmotic balance of cells. Dysfunction of these channels can cause severe diseases as Multiple sclerosis or Diabetes. Therefore, these channels are potential targets for future drugs. We investigate molecular determinants of channel blockage of druglike molecules using state-of-the-art computer simulations.
Membrane channel proteins, like ion and water channels, facilitate the permeation of water and solutes (e.g. ions) across cell membranes. They are important for many physiological processes as for example nerve conduction or the osmotic balance of cells. Dysfunction of these channels can cause severe diseases as Multiple sclerosis or Diabetes. Therefore, these channels are potential targets for future drugs. We investigate molecular determinants of channel blockage of druglike molecules using state-of-the-art computer simulations.
References:
[1] Sören J. Wacker, Camilo Aponte-Santamaria, Per Kjellbom, Soren Nielsen, Bert L. de Groot, Michael Rützler. The identification of novel, high affinity AQP9 inhibitors in an intracellular binding site. Molec. Membrane Biol. 30:246-260 (2013).
[1] Sören J. Wacker, Wiktor Jurkowski, Katie J. Simmons, Colin W. G. Fishwick, Peter Johnson, David Madge, Erik Lindahl, Jean-Francois Rolland, Bert L. de Groot. Identification of Selective Inhibitors of the Potassium Channel Kv1.1-1.2(3) by High-Throughput Virtual Screening and Automated Patch Clamp. ChemMedChem 7:1775-1783 (2012)