Inhibition Studies on hAQP1
Conformational Flooding
Chemical Flooding
Systematic Search for a "Back Door" in Acetylcholinesterase
Acetylcholinesterase (AChE) [8] is an enzyme that terminates the signal transmission at the cholinergic synapses of neurons by rapid hydrolysis of the neurotransmitter acetylcholine. The active site, a catalytic triade (SER 200, GLU 327 and HIS 440), is nearly centered in the protein. X-ray studies have shown that the active site gorge, with a depth of about 2 nm, guides the acetylcholine directly towards the catalytic triade.
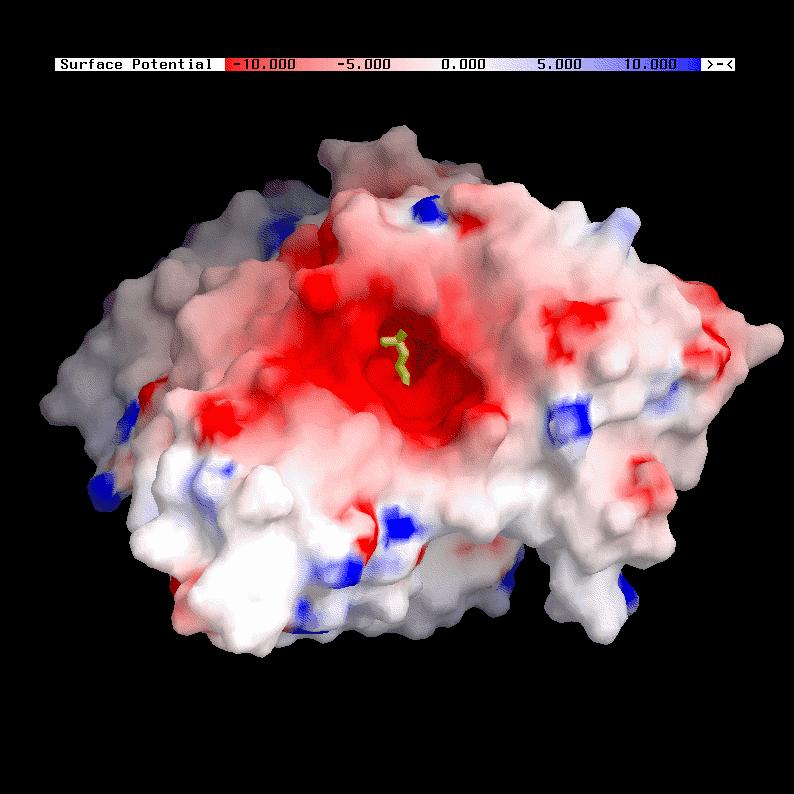 |
Due to the enzyme's high turnover number (25.000 1/s), close to the diffusion limit, and a strong dipole field, working as a kind of "vacuum cleaner" for the cationic substrate acetylcholine, it is quite unlikely that both catalysis products leave the active site through the same path by which they entered. In contrast to the well characterized entrance, the exit mechanism of the products therefore remains unknown.
 |
To allow a fast active site gorge clearance, especially for the release of the positively charged choline, a ``back door mechanism'' has been proposed. Different models for such a mechanism have been suggested [1,4]. However, all proposed backdoor models have been rejected based on biochemical experiments [7] surprisingly. Mutational studies have shown that AChE function is not affected by single mutations in the proposed back door regions.
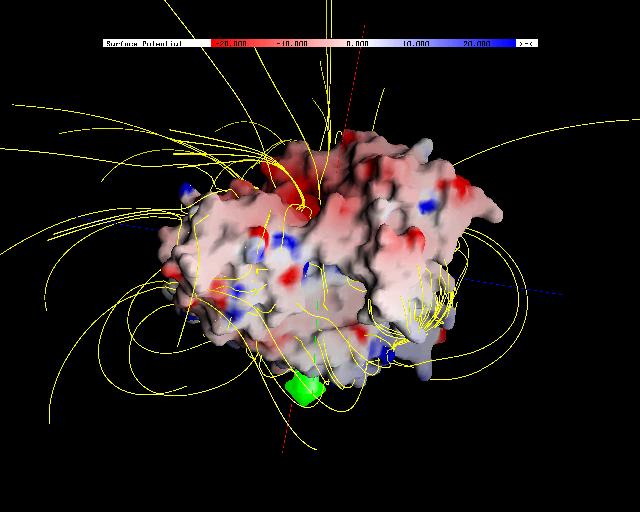
Using "Conformational Flooding" molecular dynamics (MD) simulations [2,5], we have performed a systematic search for flexible regions of the protein to find probable back doors that allow the products to exit.
Preliminary Results
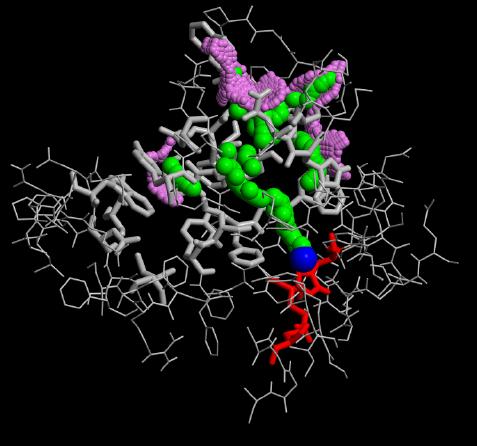
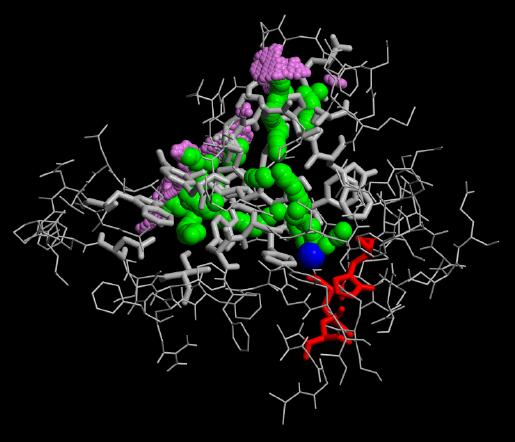
In our flooding simulations (here are shown 3 examples from different starting points for the conformational flooding and different flooding strength's) we found 2 regions (except the active site gorge) where the substrates could leave the active site.
 |
 |
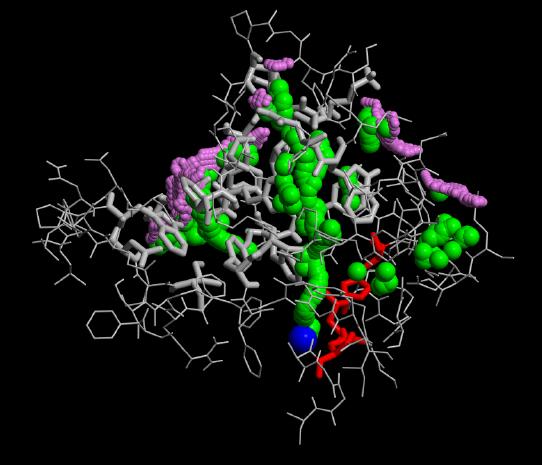 |
The grey wireframe shows the active site gorge, the catalytic triade is blue, the starting points for the pathfinder is colored red, the bulk at the end of a pathway (green) is visible in violet spheres. The green spheres show the centers of the found paths.
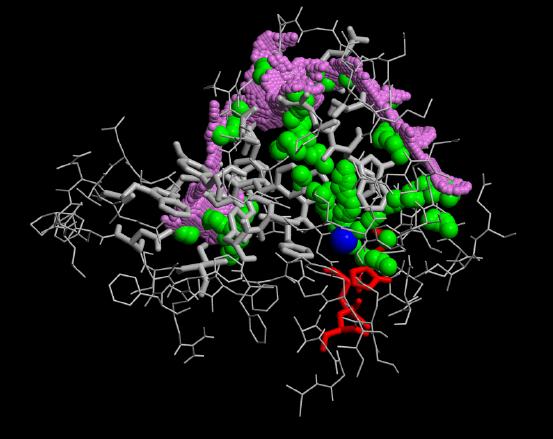
The second interesting observation arised during our calculations is that not only the conformational flooding simulations show the pathways for the probable back doors but also the conventional molecular dynamics simulation.
 |
Interim Conclusions
As can be seen from the figures in Results, obviously all paths over performed conformational flooding and conventional MD simulations are not well located. The active site gorge seems to be quite flexible in these 2 regions. This could explain that biochemical experiments realized with single mutants found the same functionality of the enzyme's catalytic power as the wild type.
References:
[1] D. van Belle, L. de Maria, G. Iurcu and S. Wodak,
J. Mol. Biol. 298(2000),705
[2] M.Eichinger, H. Grubmüller and H.Heller. User Manual for
{EGO\_VIII}, Release 1.0, 1995, electronic access:
EGO-Manual
[3] I.J. Enyedy, I.M. Kovach and B.R. Brooks,
J. Am. Chem. Soc. 120(1998),8043
[4] M.K. Gilson, T.P. Straatsma, J.A. McCammon, D.R. Ripoll, C.H.
Faerman, P.H. Axelsen, I. Silman and J.L. Sussman, Science
263(1994),1276
[5] H. Grubmüller, Phys. Rev. E52 (1995),2893
An HTML-version and a
postscript-version are available.
[6] H. Grubmüller, Solvate: A program to create atomic solvent
molecules, 1996. (electronic publication,
Solvate)
[7] C. Kronman, A. Ordentlich, D. Barak, B. Velan and A. Shafferman,
J. Biol. Chem. 269(1994), 27819
[8] D.M. Quinn, Chem.Rev. 87(1987), 955
