Inhibition Studies on hAQP1
Conformational Flooding
Chemical Flooding
Identification of the binding site for TEA in hAQP1
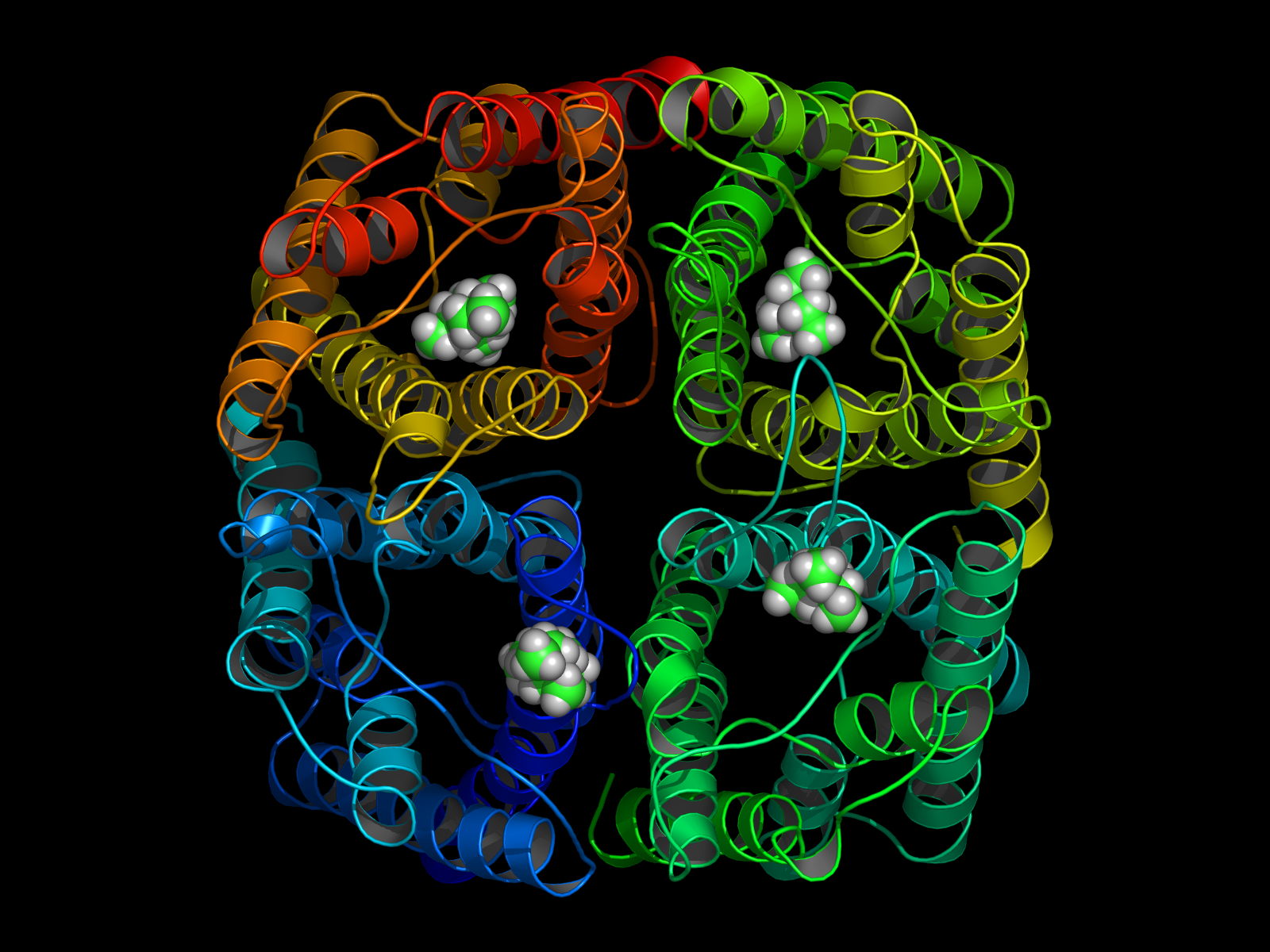
A prerequisite of performing a structure based approach in rational drug design is to know the binding site of the protein that should be targeted. In case of hAQP1 we started with a homology model, built and refined by B. L. de Groot. Furthermore, from oocyte swelling experiments it is known that TEA (tetraethylammonium) inhibits hAQP1. To identify the binding site for TEA in hAQP1 'blind docking' in the extracellular part of the pore was performed using DOCK 4.0. About 100 equally ranked poses for TEA were obtained. To refine these results, 4 representative poses were used as starting positions for molecular dynamics (MD) simulations. The MD simulations function as a 'stability test' to sort out poses which do not represent probable binding sites.
 |
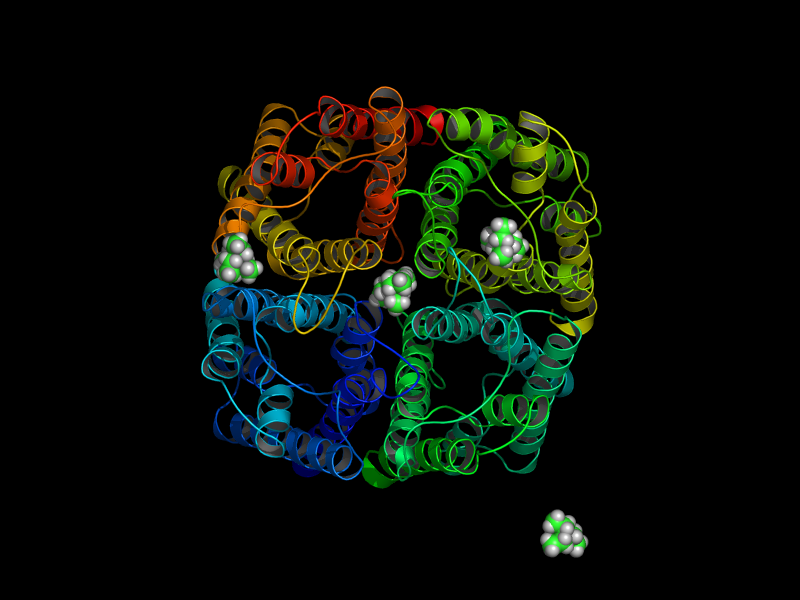 |
| starting positions | after 15ns |
|---|
In a 15ns simulation TEA stays only bound at one position. As a test, we performed a second simulation. Here, in each pore TEA was placed at the stable position specified in the 'stability test'. In this second simulation all 4 TEA remained bound. This simulation was used to characterize the binding region for TEA in hAQP1.
 |
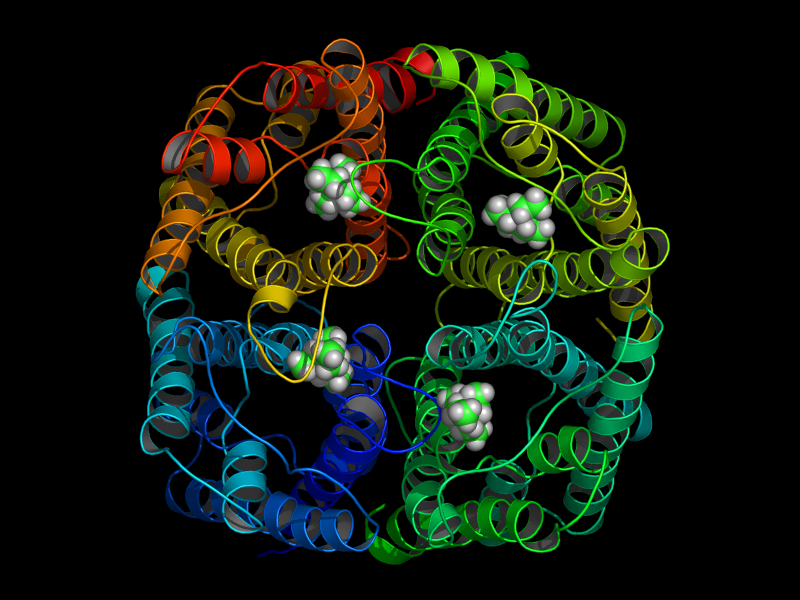 |
| starting positions | after 15ns |
|---|
For the first time a probable binding site for TEA was identified! Hence, we showed that refining docking results
with molecular dynamics simulations is a valuable method for identifying binding sites.
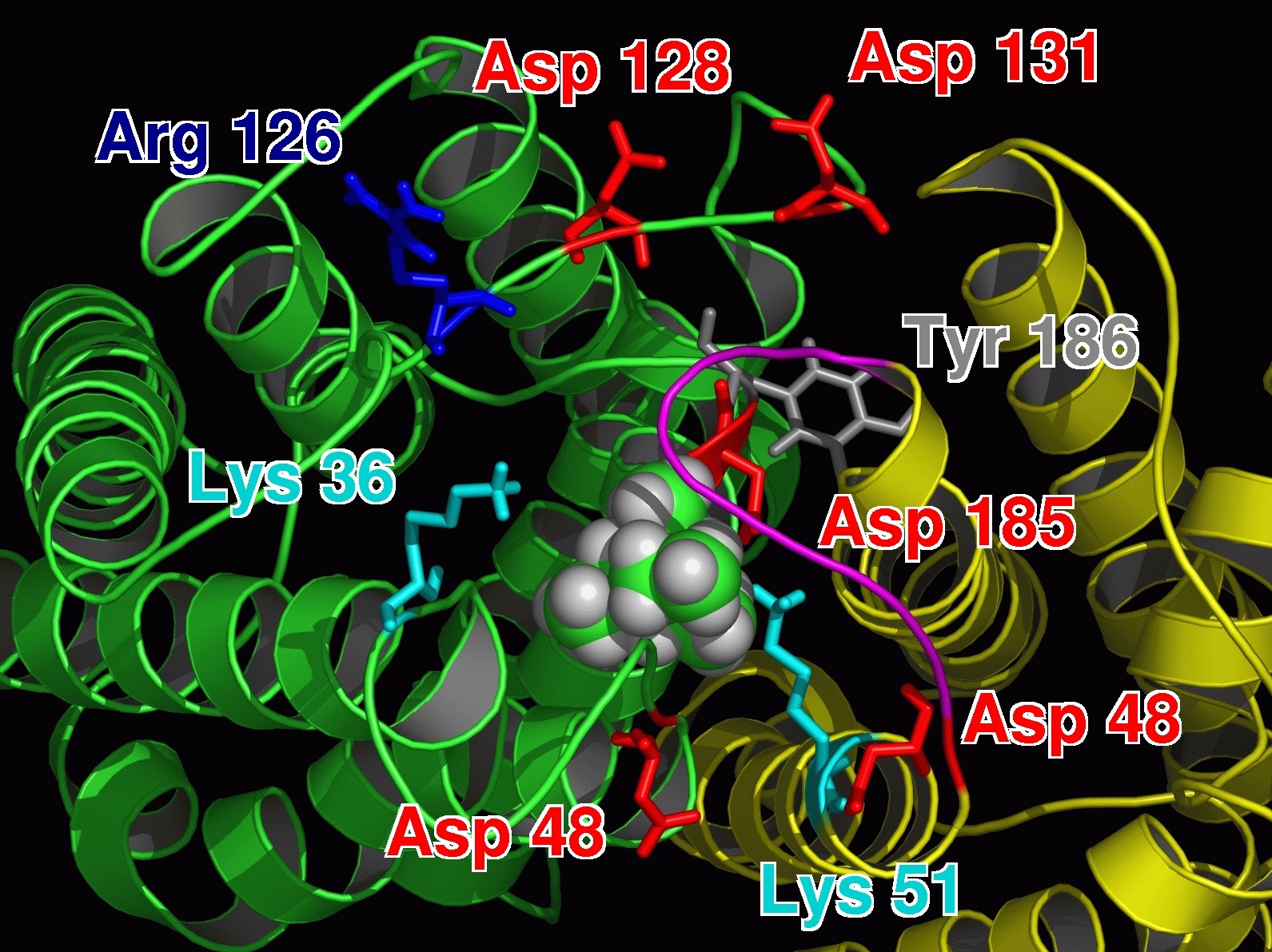 |