Inhibition Studies on hAQP1
Conformational Flooding
Chemical Flooding
'First Principles Docking'
It is a challenge to identify the binding site of a protein if 'direct' experimental data like an x-ray-, nmr- or an electron microscope structure of the protein with a bound inhibitor or an analog are not available. One way to solve this problem is to combine 'blind docking' and molecular dynamics simulations (shown here)
Here, an alternative approach is presented. 20 TEA molecules were randomly placed in the bulk water of the hAQP1 simulation system (full atomistic description of the protein, the membrane, the ions, and water molecules. See Figure, for clarity, only the protein as cartoon and TEA as cpk are shown.). Subsequently, a conventional molecular dynamics simulation was performed. During the 80ns simulation 3 binding and 2 unbinding events of TEA have been observed.
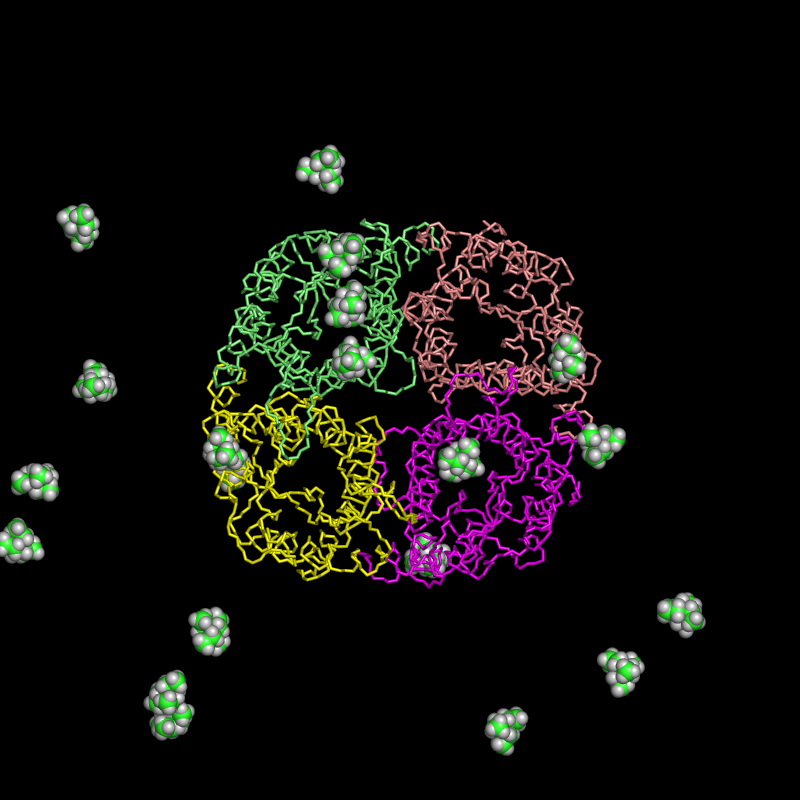 |
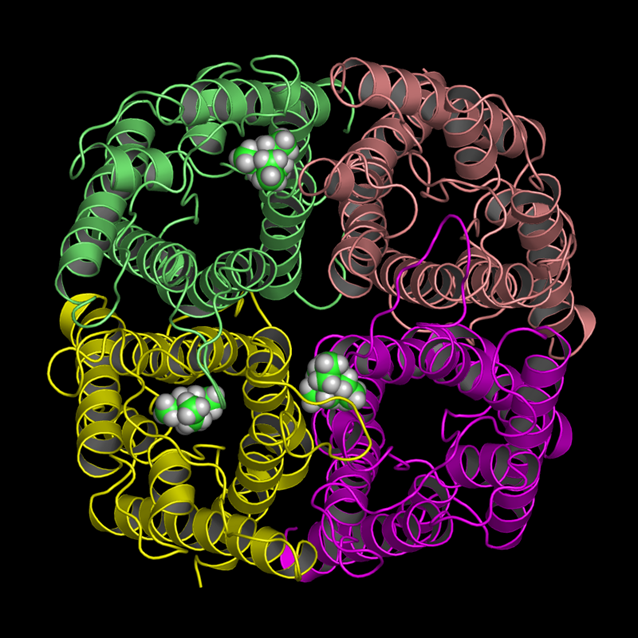 |
| starting positions | snapshot: 3 TEA bound |
|---|
All binding events started after about 5-10ns. Two binding events lasted for about 15-20ns
(snapshot: 3 TEA bound, left side). The third one lasted until the end of the 80ns. Here, TEA bound at the same
binding site that already has been identified previously.