Inhibition Studies on hAQP1
Conformational Flooding
Chemical Flooding
Predicting Unimolecular Chemical Reactions: Chemical Flooding
The calculation of reaction pathways, energy barriers and reaction rates for chemical reactions of small molecules is a routine task in todays theoretical quantum chemistry. Established methods exist for tasks like (a) minimization of educt states, (b) exploring potential energy hypersurfaces, (c) detecting transition and product states, (d) finding reaction pathways connecting the educt, transition and product state, and (e) calculating the reaction rates by associated quantum statistical mechanical approaches.
Assuming a given educt state, all these methods require additional knowledge on the reaction pathway, putative transition states or the product state.
Recently, a force field based method, 'conformational flooding'[4] has been developed to predict structural transitions of macromolecules. Here we extend that method towards the calculation of chemical reactions using density functional theory. Accordingly, that extension, called 'chemical flooding', should enable one to predict in an unbiased manner reaction pathways, transition states and accessible product states for dissociation reactions of small molecules. 'Chemical Flooding' also provides estimates for activation energies, which can and should subsequently be refined with already established methods.
|
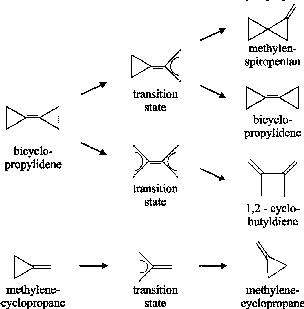
As a test case, we studied the known dissociation reaction of two molecules, bicyclopropylidene (BCP) and methylenecyclopropane (MCP)[1].
Results
|
Our 'chemical flooding' simulations predict three reaction pathways
for BCP (Figs. 1-4) and one for MCP (Fig.
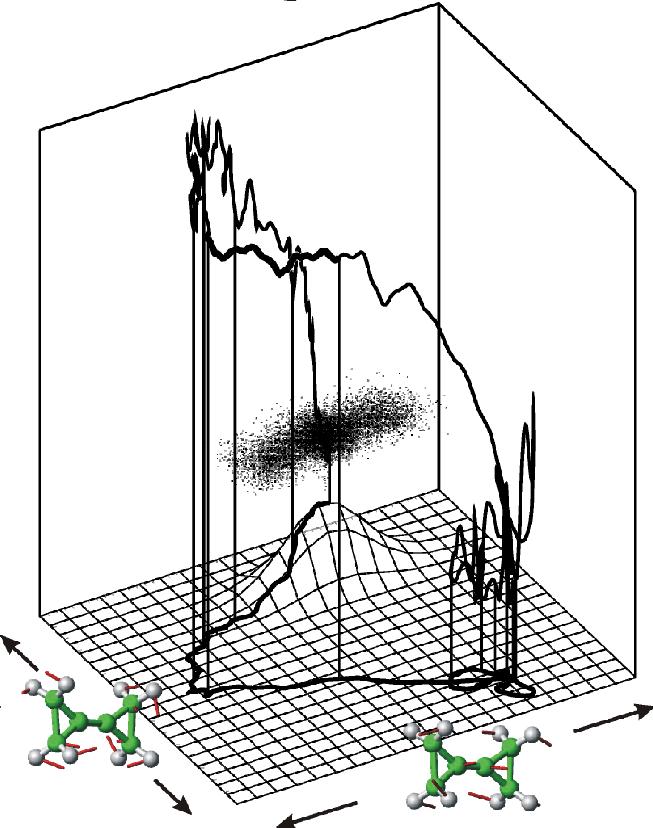
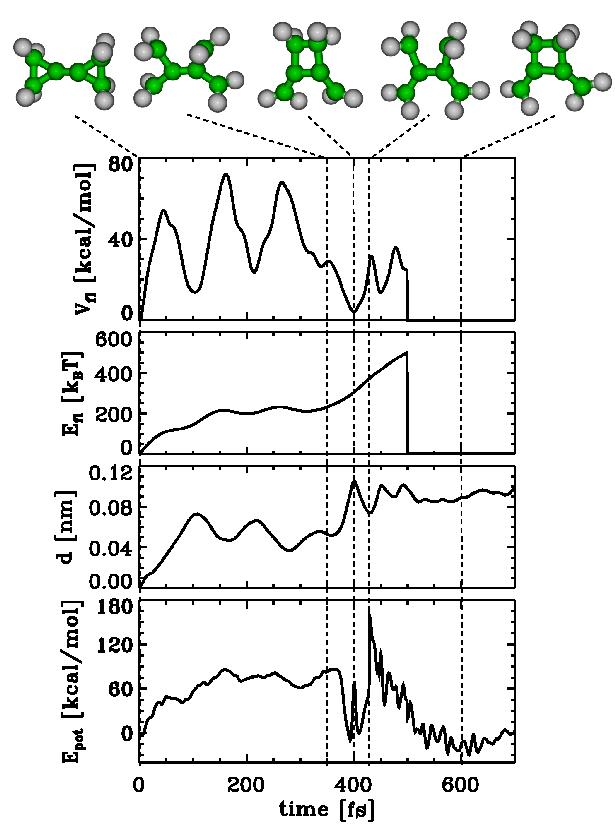
5). Fig. 1 illustrates a typical case. The bottom curve
shows the predicted BCP to methylenespiropentane (MSP)
reaction pathway in the configurational space spanned by the two
essential degrees of freedom which contribute most to the educt
equilibrium fluctuations. The vertical axis and the upper trajectory
show the potential energy along the pathway. The main barrier is about
40-50 kcal/mol. Also shown as a mesh-plot is the used flooding
potential Vfl.
|
|
|
|
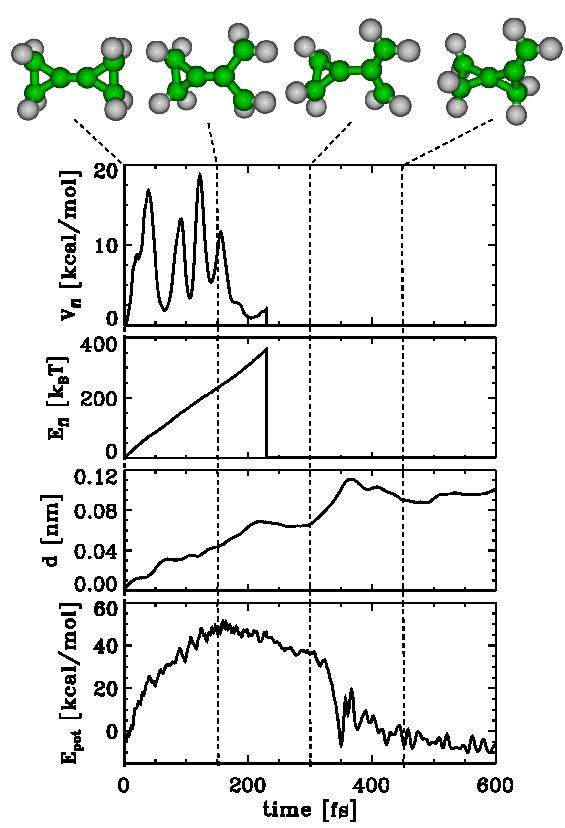
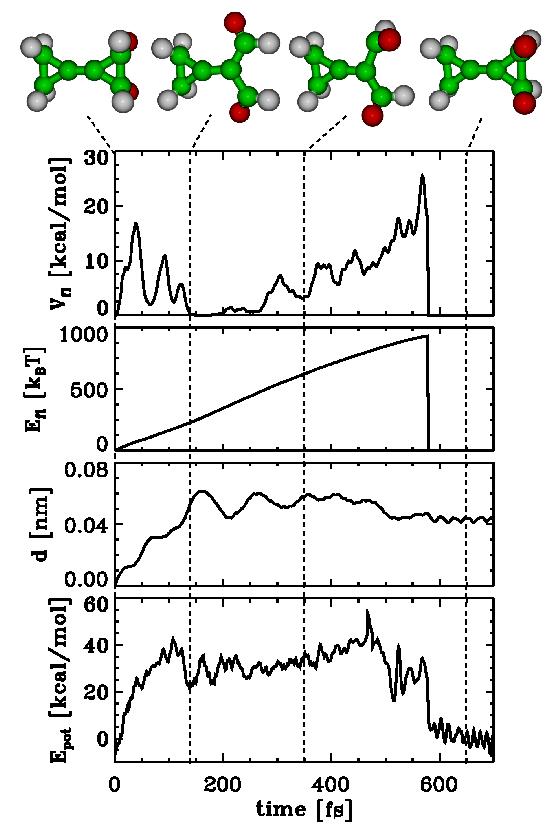
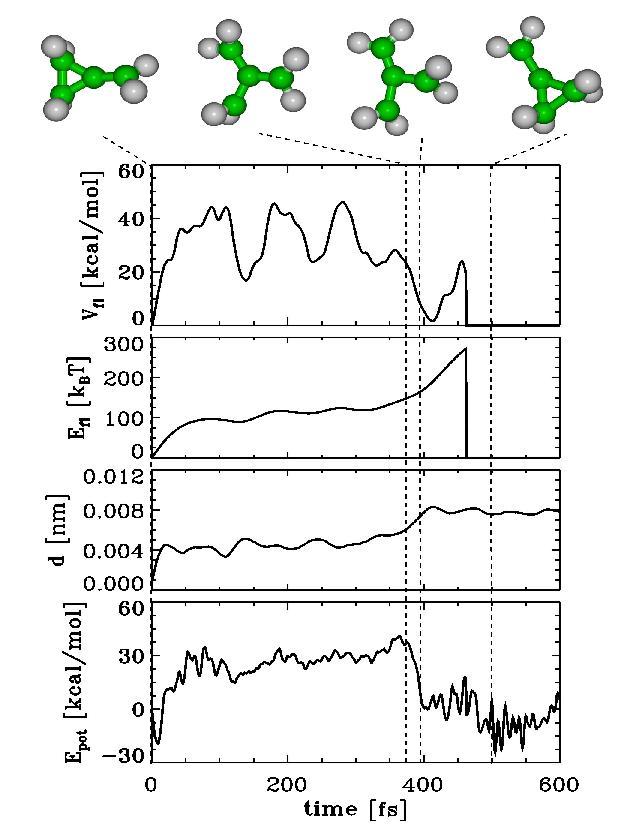
| Figs. 2-5 quantify our results for BCP (Figs. 2-4) and MCP (Fig. 5), respectively. Shown are the potential Energy Epot, the root mean square deviation (rmsd) d with respect to the starting structure of the respective chemical flooding run, the flooding strength Efl, and Vfl, the actual value of the flooding potential. |
|
|
|
| As can be seen in Figs. 2-5, an increasing flooding strength E fl drives the molecule over a barrier in Epot. The distance travelled along the pathway is monitored via the rmsd value d. The thermal vibrations of the molecule show up as fluctuations in V fl as well as Epot. |
|
|
|
| For the activation energies we estimate for the reactions BCP to MSP Ea ~ 40-50 kcal/mol (Fig. 1,2), BCP 'hflip' reaction Ea ~ 30-40 kcal/mol (Fig. 3), BCP to 1,2 - cyclobutyldiene (CBD) Ea ~ 80-100 kcal/mol (Fig. 4) and MCP reorganisation Ea ~ 40-50 kcal/mol (Fig. 5). This compares favourably with experimental values for BCP to MSP (39,2 kcal/mol) [2] and MCP to MCP (40,4 kcal/mol). |
|
|
|
Conclusions
|
|
|
|
References:
[1] A. de Meijere, S. Kozhushkov, and A. F. Khlebnikov. Topics in Current Chemistry , Vol. 207, Springer-Verlag, Berlin, Heidelberg, 2000.
[2] W. R. Dolbier, K. Akiba, J. M. Riemann, C. A. Harmon, M. Bertrand, A. Bezaguet, and M. Santelli, J. Am. Chem. Soc., 93(16):3933-3940, 1971
[3] M.Eichinger, H. Grubmüller and H.Heller. User Manual for {EGO\_VIII}, Release 1.0, 1995, electronic access: EGO-Manual
[4] H. Grubmüller, Phys. Rev. E52 (1995),2893
An HTML-version
and a
postscript-version
are available.
[5] J. Hutter et al. CPMD version 3.0 manual. IBM Research Division, Zürich research Lab. MPI für Festkörperforschung, Stuttgart, 1995-1998
